Command Palette
Search for a command to run...
材料AIは「説明可能な時代」へと向かっている:日本の研究チームが高次元分光法のブラックボックスを解明し、新素材発見のための重要な特徴を特定した。

近年、材料科学分野における機械学習の応用が大きな注目を集めている。その応用範囲は、初期の構造・物性スカラー予測(バンドギャップエネルギー、点欠陥形成エネルギー、融点など)から、より複雑な高次元物理量のモデリングへと徐々に拡大してきた。中でも最も挑戦的な方向性の一つが、材料スペクトルの予測と解析である。
誘電関数、スペクトル(吸収、反射、発光)、電子状態密度、フォノン状態密度などのスペクトルデータは、材料の理解と設計において極めて重要です。しかし、スカラー特性と比較して、高次元スペクトルデータは、出力次元が大きく、構造が複雑で、物理的制約が強いため、従来の機械学習手法では精度と解釈可能性を同時に達成することが困難です。深層学習モデルはある程度スペクトルを予測できるようになりましたが、解釈可能性の欠如が依然として大きなボトルネックとなっており、材料設計におけるさらなる応用を阻んでいます。
この文脈では、日本の東京理工大学の研究チームは、材料科学における高次元スペクトルデータを処理できる深層学習モデルの解釈方法を提案した。研究者らは、2,681種類の酸化物、カルコゲニド、および関連化合物の光吸収スペクトルに関する第一原理計算のデータセットを構築した。標準的な密度汎関数計算と比較すると、計算結果はスペクトルの開始エネルギーと形状を補正した後、報告されている実験スペクトルとの一致が大幅に向上したことが示された。
研究者らはまた、このデータセットとALIGNNアルゴリズムを用いて、高精度な光吸収スペクトル予測モデルを開発した。特徴抽出とクラスタ分析を組み合わせることにより、光吸収の開始エネルギーと強度を主に決定する主要な元素の種類とその配位環境を抽出することに成功した。
関連研究の成果は、「深層学習に基づく高次元データからの有望な材料グループと共通特徴の抽出:無機結晶の光スペクトルの事例」と題され、Advanced Intelligent Discovery誌に掲載された。
研究のハイライト:
* 本研究では、高次元スペクトルデータの特徴抽出とクラスタ分析による材料分類手法を提案し、それによって潜在的な材料グループとその共通の特徴を抽出する。
* 本研究で開発された第一原理計算データセットと機械学習モデルは、将来の材料発見および材料情報学研究において重要な役割を果たすことが期待される。
本研究で提案する手法は幅広い適用性を持ち、様々なスペクトルデータの分類や解釈に利用できます。その適用範囲は無機結晶の光吸収スペクトルに限定されません。

用紙のアドレス:https://advanced.onlinelibrary.wiley.com/doi/10.1002/aidi.202600007
高スループットの第一原理計算を用いて構築されたデータセット
研究者らはまず、Materials Project データベースから、以下の条件を満たす酸化物、カルコゲニド、および関連材料をスクリーニングした。(1) 材料には、O、S、Se のいずれかの元素が少なくとも 1 つ含まれており、その酸化数は必ずしも −2 ではない。(2) 材料には、H、ハロゲン、希ガス、Mn–Ni、Tc–Rh、Os–Ir、Po、ランタニド (La と Ce を除く)、アクチニドなどの元素が含まれていない。(3) 材料はスピン偏極を示さない。(4) 空間群 P1 および/または元の単位格子に 40 個を超える原子を含むシステムは、計算コストが高いか結晶構造に不確実性があるため除外された。
第一原理計算に使用された材料の総数は9,808であり、計算データベースは下図に示すプロセスに従って構築された。
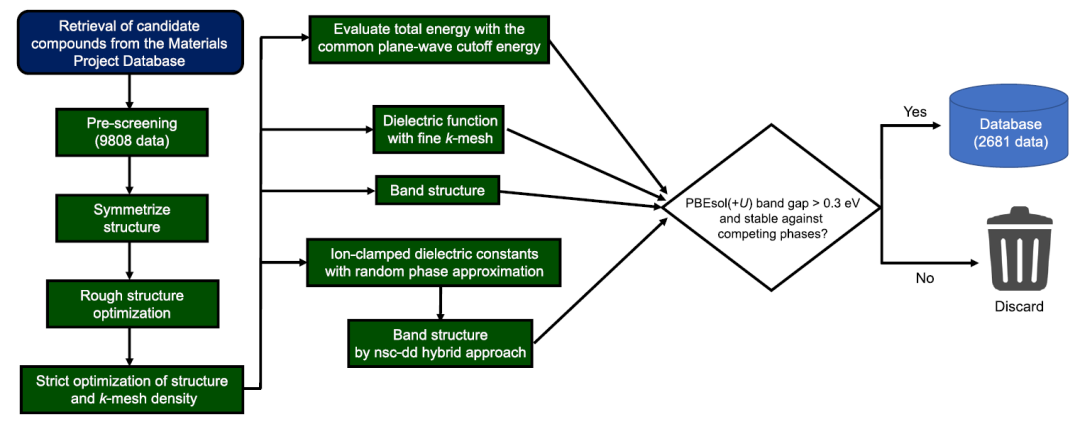
図に示すように、この計算プロセスは非常に複雑です。一貫性と計算リソースの効率的な使用を維持しながら、高スループットの計算を実現するために、研究者たちは独自に開発したプログラムを使用し、pymatgen、FireWorks、Custodian、atomate、viseといったツールを活用してプロセスを自動化した。すべての第一原理計算はVASPソフトウェアパッケージを使用して実行されました。このワークフローでは、PBEsol(+U)計算を使用して光吸収スペクトルと化合物形成エネルギーを生成し、nsc-dd混合汎関数とPBEsol(+U)計算を使用してバンド構造を取得します。
機械学習データセットに関して、研究者らは、(1)ローカルデータベース内の競合相に対して不安定な材料、および(2)PBEsol(+U)バンドギャップが0.3 eV未満の材料を除外した。最終的に残った材料の数は2681であった。
光吸収スペクトルに基づくALIGNNモデルの構築
機械学習モデルの構築と予測精度
モデルレベルでは、本研究では、高次元の光吸収スペクトルをモデル化するための主要な予測フレームワークとして、ALIGNN(Atomistic Line Graph Neural Network)を使用しています。従来の結晶グラフ畳み込みネットワーク(CGCNN)と比較して、ALIGNNの核心的な利点は、「原子グラフ+結合線グラフ」という2つの表現を同時に導入することで、3体角情報を明示的に符号化し、局所的な構造環境をより精緻に表現できる点にある。下図の上部は、ALIGNNアーキテクチャの概略図である。
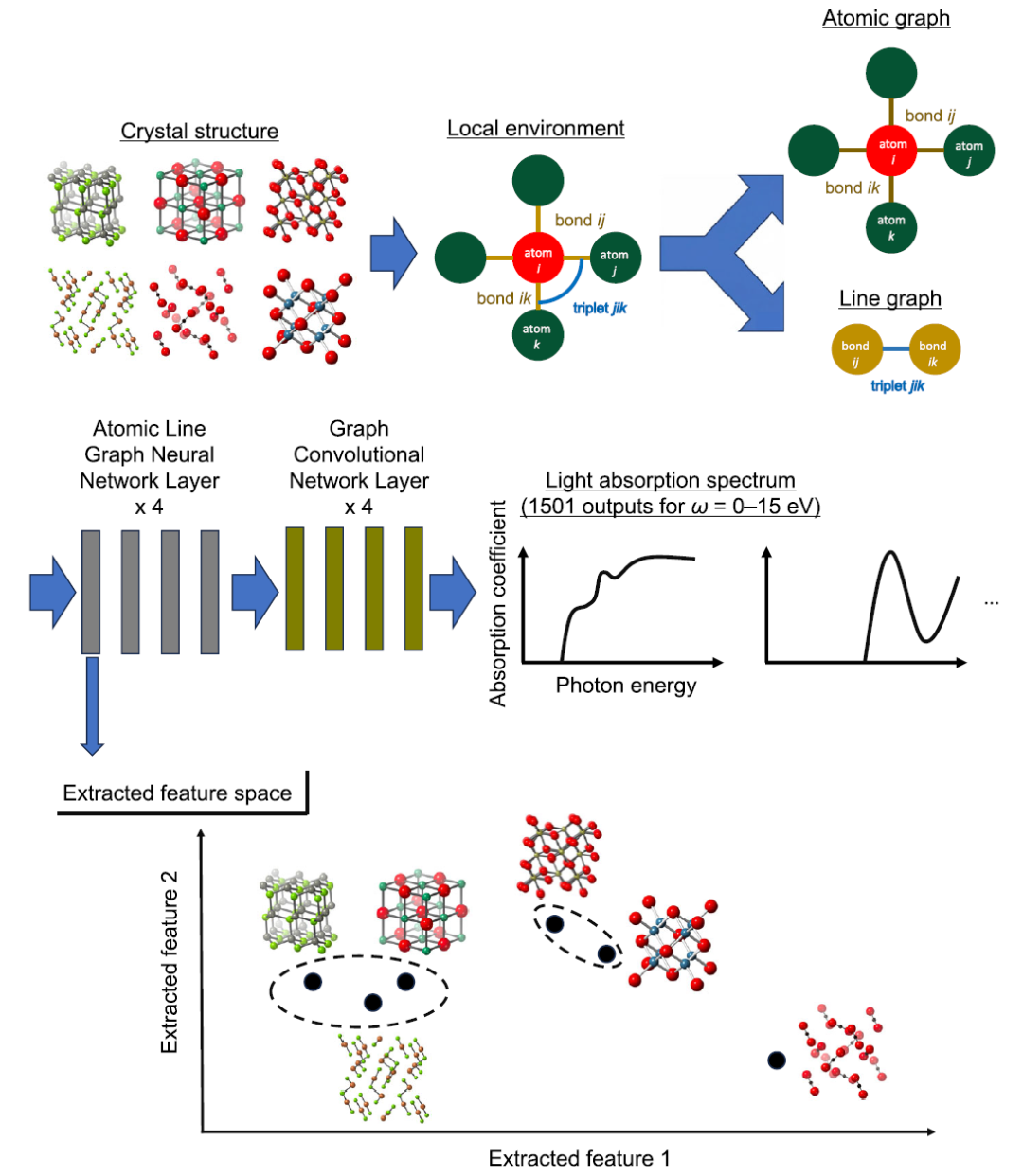
このフレームワークでは、原子はノード、原子間結合はエッジであり、エッジ間の関係はさらに線グラフとして構築され、それによって結合角の情報が学習可能な構造的特徴に変換される。この設計により、モデルは2つの物体間の距離情報を捉えるだけでなく、3つの物体間の相互作用を特徴づけることも可能になり、結晶の物理的挙動により近いものとなる。
特徴抽出とクラスタリング
研究者らは、最適化されたモデルのALIGNNの第1層から特徴を抽出し、各材料のすべての原子サイトの特徴ベクトルを平均化してから、階層的クラスタリング分析を実行しました(上の図の下半分参照)。この方法の目的は、入力特徴(元素組成や、隣接原子の数、原子間距離、結合角などの原子配位特徴など)と出力特性(光吸収スペクトル)の両方において類似性を示すグループに材料を分類することです。
下図は、階層的クラスタリングによって得られた96グループの光吸収スペクトルを示している。各クラスター内のスペクトル形状は確かに類似しており、本研究におけるクラスタリング手法の有効性が確認された。

結果:物質集団構造と物理的メカニズムの解釈可能な抽出が達成された。
材料科学における高次元スペクトルデータを処理する新しい深層学習モデルの能力を検証するため、研究者らは一連の実験を行った。
予測性能
予測性能に関して言えば、ALIGNNモデルはテストセットにおいて全体的に高い精度を示しました(下図参照)。約75%の物質吸収スペクトル予測の平均絶対誤差(MAE)は0.14未満であり、このモデルが複雑なスペクトル形状を良好に再現できることを示している。
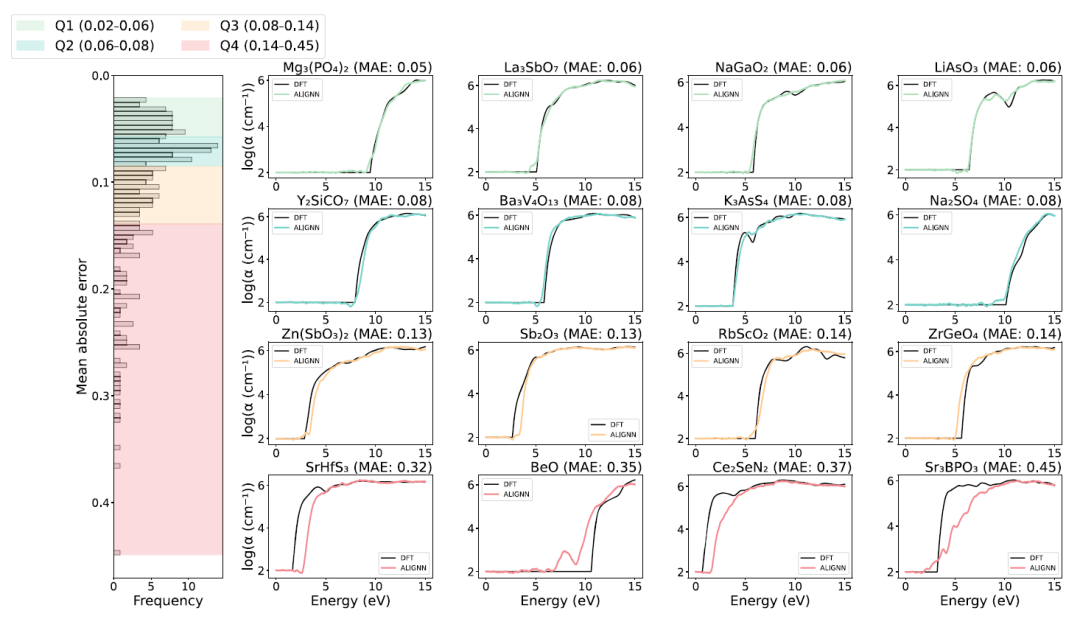
上の画像の右側のパネルは、各四分位で誤差が最も大きい4つの材料の予測結果を示しています。最初の3つの四分位の材料については、ALIGNNの予測結果(色付きの曲線)は、第一原理参照計算結果(黒色の曲線)とよく一致しています。しかし、第4四分位の一部の化合物は、光吸収スペクトルの開始位置に大きなずれが見られます。これらの外れ値サンプルは、主にその特異な電子構造と、トレーニングデータセットに類似の構造を持つ材料が不足していることが原因で、予測性能が低くなっています。
光吸収スペクトルの開始位置を捉える能力
MAEはスペクトル範囲全体をカバーするグローバルな指標ですが、研究者らはさらに、モデルが局所的なスペクトル開始エネルギーを正確に再現できるかどうかを検証しました。下の図はパリティプロットを示しています。これは、第一原理計算とALIGNN予測において、log₁₀ α(ω)が初めて2.5を超えるときの最低光子エネルギーを比較したもので、αは吸収係数を表します。
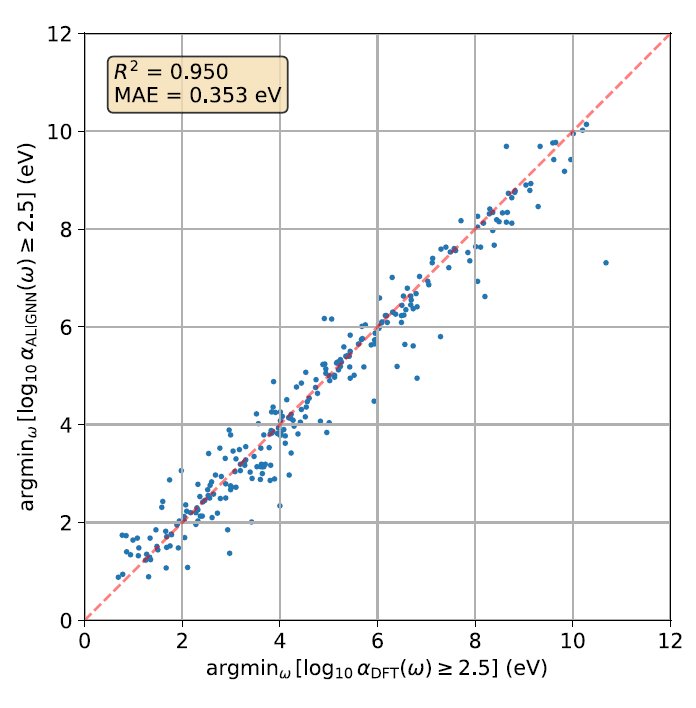
結果によると、初期エネルギーの予測R²は0.950、MAEは0.353 eVであり、ALIGNNモデルが光吸収スペクトルの開始位置を正確に捉えることができることを示している。
解釈可能性の分析
解釈可能性分析の観点から、研究者らはALIGNNの第1層から特徴表現を抽出し、材料の階層的クラスタリングを実行した結果、96の材料グループが得られた。その結果、…同一クラスター内の物質は、スペクトル形状、特に吸収開始位置と吸収端の傾斜において高い一貫性を示し、顕著な共通性を示している。これは、モデルが初期層におけるスペクトル関連構造特性を学習したことを示唆している。
さらに事例研究を行うと、異なる材料グループ間で明確な物理的差異が明らかになります。例えば、グループ74は一般的に、広いバンドギャップとスペクトル変曲点付近で高い吸収係数を示します。図aは、このグループのすべての材料がVまたはCrのいずれかを含み、他の陽イオンは主にアルカリ金属であることを示しています。これらの材料は主にVO₄³⁻、CrO₄²⁻、またはCr₂O₇²⁻の形で存在し、陽イオンは四面体配位環境にあります。
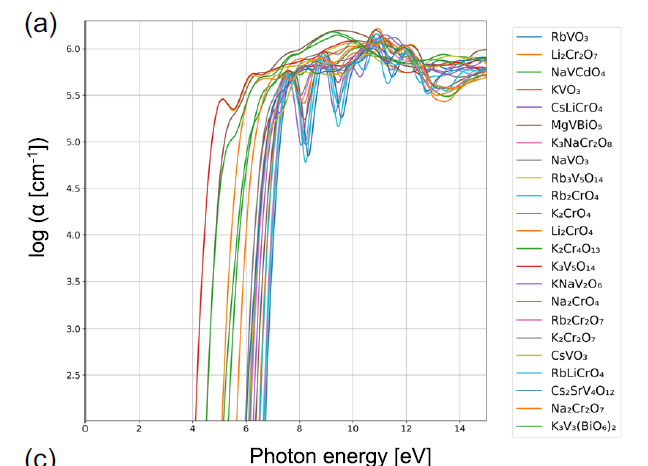
クラスター74に属する物質の光吸収スペクトル。ここでαは吸収係数を表す。
研究者らは、Matminerに実装されているCrystalFingerprintNNを用いて、クラスター内の各材料における陽イオンサイトの四面体配位指数を計算し、すべての陽イオンサイトの最大値の分布を分析した。下の図bに示すように、ほとんどの材料は確かに四面体配位サイトを有している。
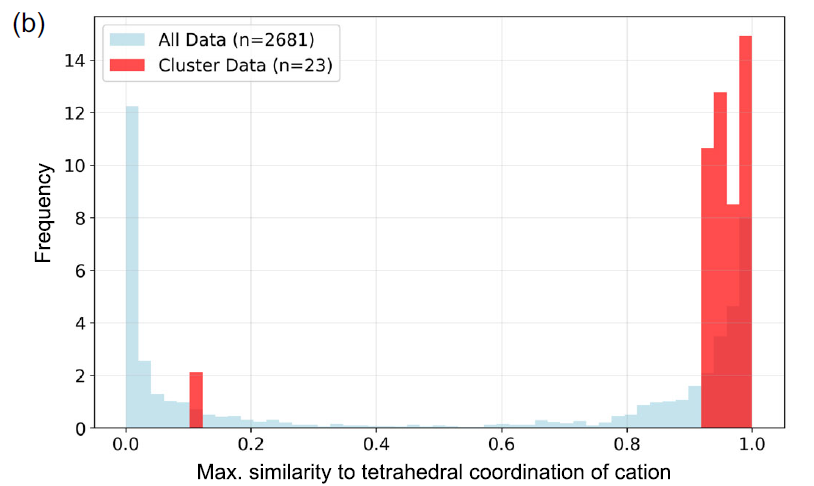
74番目の材料クラスター(赤)とデータセット全体(青)との間の四面体配位類似性の分布。
電子状態密度の観点から見ると、伝導帯底(CBM)付近にVd状態またはCr-d状態に起因する鋭いピークが観測される。V⁵⁺およびCr⁶⁺の高価数状態は、光遷移に利用できる多数の非占有電子状態を提供する。したがって、固体化学および物理学の観点から、これらのバナジン酸塩、クロム酸塩、および二クロム酸塩が高い光吸収係数を持つことは妥当である。
モデルクラスタリング結果から化学反応機構を推論するこのプロセスは、機械学習の結果をブラックボックス的な予測から材料設計のための貴重な知識源へと変革します。さらに、この研究では、生のスペクトルデータに基づく直接クラスタリングの結果と比較したところ、類似のスペクトルを識別することはできたものの、明確な化学構造グループを形成するのに苦労し、結果として材料の種類が大きく混在してしまうことがわかりました。これは、一貫した構造-物性表現を実現する上で、ALIGNN特徴空間が持つ利点をさらに示しています。
結論
本研究の意義は、高精度な光吸収分光予測モデルを構築した点だけでなく、より重要な点として、「深層学習による表現学習」と「材料物理学的解釈」を組み合わせた方法論的枠組みを提案した点にある。ALIGNNモデルと階層的クラスタリング分析を組み合わせることで、高次元スペクトルデータから材料の共通法則を抽出することが可能となり、機械学習モデルが結果を予測するだけでなく、その結果の根底にある構造や電子的な起源を明らかにすることも可能になる。
理想的には、励起子効果、フォノン補助電子遷移、および欠陥のスペクトル特性をそれぞれ再現するために、電子-正孔相互作用、電子-フォノン結合、および点欠陥の影響を考慮に入れるべきである。しかし、これらの効果を含めたハイスループットな第一原理スペクトル計算は計算コストが高すぎるため、本研究では実現できなかった。それでもなお、より高精度な多体計算手法と機械学習モデルのさらなる統合により、この種の研究は材料発見においてより中心的な役割を果たすことが期待され、材料設計を経験主導型アプローチから、データ駆動型アプローチとメカニズム駆動型アプローチを統合した新たな段階へと押し上げるだろう。