Command Palette
Search for a command to run...
科学者たちは、ベイズ最適化フレームワークを用いてガリウム含有材料をリバースエンジニアリングすることで、独自に新しい材料を生成した。最適化の結果は、独自性と新規性を示している。

現代の半導体産業では、材料性能の限界が絶えず高次元へと押し上げられています。高効率太陽電池から高輝度発光ダイオード(LED)、そして高周波通信や量子情報システムに至るまで、ほぼすべての主要技術は、その中核となるコア機能に依存しています。材料の電子構造を精密に制御すること、特にバンドギャップを精密に設計すること。しかし、この目標は従来の材料科学の体系においては長らく達成が困難であった。
その理由は、物質の電子特性は単一の元素だけで決まるのではなく、複雑な化学結合、結晶構造、電子軌道の混成、そして複数の元素の相乗効果によって影響を受けるからである。数ある材料系の中でも、ガリウム系半導体は独特な位置を占めている。ガリウムは、その優れた化学的多様性と多価状態特性により、広いバンドギャップから狭いバンドギャップまで、調整可能な幅広い電子特性を示すことができる。
ガリウム含有化合物は、高効率太陽電池、高輝度LED、高周波通信機器といった主要な光電子技術やエネルギー変換技術の重要な基盤となっています。また、柔軟性、生体適合性、埋め込み性を備えた電子システムの有望な候補材料としても注目されています。しかし、数十年にわたる研究にもかかわらず、特定の電子特性をターゲットとした新規ガリウム含有材料の発見は、依然として経験的な探索に大きく依存しています。これは主に、部品の設計空間が膨大であることと、第一原理に基づく計算の計算コストが高いことによって制限される。
こうした背景のもと、フリンダース大学が主導し、アラブ首長国連邦のハリファ大学と共同で研究を行ったチームは、化学的合理性を維持しながら、あらかじめ定義された電子特性を持つガリウム系部品の逆設計を可能にする、機械学習を活用したベイズ最適化(BO)フレームワークを提案した。
この統一フレームワークの助けを借りて、このシステムは、新規かつ化学的に有効なガリウム含有材料を自律的に生成し、0.5~3.5 eVの範囲で調整可能なバンドギャップを実現できる。このエネルギー範囲は、太陽エネルギー、フォトニクス、パワーエレクトロニクスなどのアプリケーションにとって非常に重要です。ベイズ最適化プロセスは、最も「望ましい改善」が得られる領域へと探索を適応的に導くことができます。最適化された分析結果によると、生成された材料はトレーニングデータに対して100%の独自性と新規性を持ち、1.5~2.5 eVのバンドギャップ範囲でSMACTの有効性が大幅に向上しています。
関連する研究成果は、「ベイズ最適化による、目標とするバンドギャップを持つガリウム含有半導体の発見」と題され、ACS Publicationsに掲載された。
研究のハイライト:
この新しいフレームワークは、現実的な化学的制約の下で逆材料設計を加速させることができ、DFT(密度汎関数理論)に基づく従来のスクリーニング方法に代わる選択肢を提供する。
* 新しいフレームワークは、化学的に妥当な領域を効率的に網羅するだけでなく、既存のデータベースと比較して高い新規性と構成要素の多様性を維持しています。
この研究は、従来の静的な特性予測の限界を打ち破り、半導体発見をデータ駆動型の生成研究パラダイムへと推進するものです。

用紙のアドレス:
https://pubs.acs.org/doi/10.1021/acsmaterialslett.5c01482
データセット:実世界の材料データベースから化学学習空間を構築する
本研究では、NOMADおよびMaterials Projectのデータベースを用いてモデルを訓練した。データには、材料の化学組成と、それに対応する実験的なバンドギャップ値が含まれています。例えば、Ga₄P₄、GaAs、GaN、Ga₂O₃など。初期データセットには、2,530種類の材料組成とそのバンドギャップ記録が含まれています。
データ品質を確保するため、「composition」列または「band_gap」列に欠損値があるサンプルは削除しました。非物理的または負のバンドギャップデータも削除し、重複レコードも削除した結果、最終的にモデリングに使用できる有効なコンポーネントが 1,578 個残りました。さらに、化学式文字列は pymatgen パッケージを使用して標準化し、化学的に同等の用語を統合しました。バンドギャップの単位は、ジュールから電子ボルト (eV) に統一的に変換しました。前処理済みのデータセットでは、バンドギャップは 0.0 ~ 5.92 eV の範囲で、平均は約 1.8 eV、標準偏差は 1.6 eV でした。
この研究ではさらに材料組成を精査し、あらかじめ定義された原子番号の元素を含む化合物のみを残すことで、ガリウム系材料系に焦点を当てた研究を実施した。また、以下のようないくつかの追加機能も構築した。
* 各化学式に含まれる元素の数
* 化学式文字列の長さ
ガリウムの有無を示す二値指標
データセットは、化学的に類似した化合物が異なるデータセットに同時に出現することを避けるため、「成分レベル」で分割され、8:2の比率でトレーニングセットとテストセットにランダムに分割された。また、異なるデータ分割条件下におけるモデルの堅牢性を評価するために、5分割交差検証も用いられた。
フレームワーク:機械学習とベイズ最適化の共同設計
本研究では、化学的制約を考慮したベイズ最適化(BO)フレームワークを提案する。下図に示すように、まずガリウム系複合材料データセットで学習させた勾配ブースティング回帰モデルを用いて材料のバンドギャップを予測します。次に、ベイズ最適化を用いて制約付き組成空間を反復的に探索します。最後に、生成された候補材料をSMACTおよびpymatgenツールを用いて化学的妥当性、新規性、独自性についてスクリーニングし、これまで探索されていなかった最高の性能を持つガリウム系複合材料を特定します。
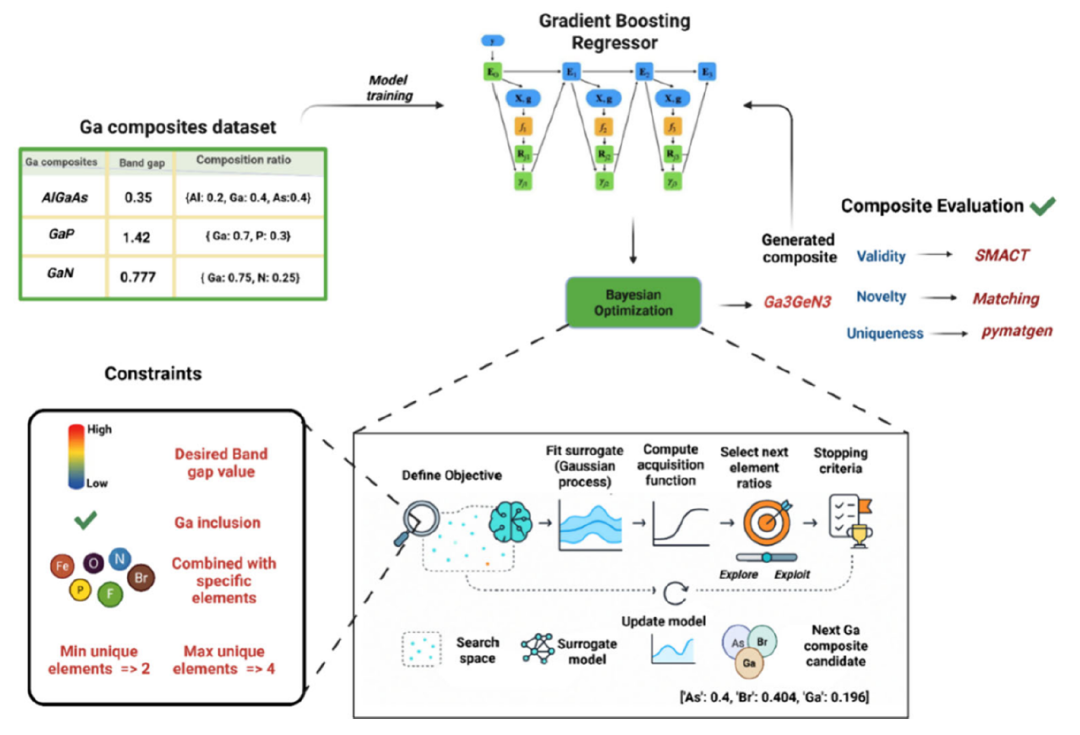
予測モデル層
本研究では、線形モデル、サポートベクター回帰、ランダムフォレスト、勾配ブースティング、K近傍法(KNN)を含む8つの機械学習回帰アルゴリズムを体系的に評価した。その結果、非線形モデルが全体的に線形モデルを大きく上回る性能を示し、材料組成とバンドギャップの間に強い非線形関係が存在することが明らかになった。KNNモデルが最も優れた性能を示し、R²値は0.812を達成しただけでなく、誤差指標においても他のモデルを上回った。
候補となるモデルの中から、最終的にKNNがベイズ最適化における代理モデルとして選択された。その理由は、優れた局所補間能力を持ち、様々なランダム分割条件下でも安定した性能を維持するためである。ツリーベースのアンサンブルモデルとは異なり、KNNは構成要素の特徴空間における近傍関係を保持することができ、これは類似した元素比率を持つ材料間の類似性を識別する上で非常に重要である。
ベイズ最適化のシナリオでは、この「局所保存機能」は特に重要です。なぜなら、最適化探索は、既知の高品質候補の近傍にある潜在的な領域に焦点を当てることが多いからです。したがって、KNNの非パラメトリックかつ局所適応的な特性は、疎にサンプリングされた材料空間において高い計算効率を維持しながら、最適化アルゴリズムに、よりスムーズで信頼性の高い探索ガイダンスを提供することができます。
ベイズ最適化モジュール
このBOワークフローは、KNNサロゲートモデルを利用して、ターゲットバンドギャップ内のガリウム含有成分の探索を誘導します。「期待される改善」取得機能を用いることで、「探索」と「活用」のバランスが取れ、ガリウムを中心とした組成空間における候補となる化学量論比が生成される。
このシステムでは、いくつかの制約が設けられており、例えば、各構成要素は最大4つの元素を含み、ガリウム含有量の最小要件を満たさなければならない。これは、候補となる材料がガリウムをベースとした研究テーマとの関連性を維持するためである。
化学的制約のあるフィルター層
生成されたすべての候補材料は、電荷バランス、妥当な酸化状態、電気陰性度の一貫性などの制約を含め、SMACTツールを使用して検証されなければなりません。これにより、生成された材料が数学的な空間で有効であるだけでなく、化学的に実現可能であることが保証されます。
さらに、このフレームワークは説明可能な人工知能(XAI)手法を取り入れており、SHAPを利用してモデルの意思決定ロジックを分析することで、材料予測を「ブラックボックス」から「説明可能なシステム」へと変革します。
現実的な化学的制約の下での材料逆設計の加速
研究者たちは、モデルの性能、構造的特徴、解釈可能性、および化学的妥当性を評価・分析するために、一連の実験を設計した。
モデル性能評価
モデル性能評価に関して、KNNモデルは交差検証において安定性を示し、R²は約0.60±0.07、RMSEは約1.02 eVであり、このモデルが疎な化学空間において優れた汎化能力を持っていることを示している。
以下の特徴重要度分析に示すように、融点、電気陰性度範囲、および電気陰性度偏差は、バンドギャップ予測に影響を与える重要な要素であり、材料の結合強度と電荷移動挙動に密接に関係しています。電気陰性度の差が大きくなるにつれてバンドギャップは減少する傾向があり、一方、融点と凝集エネルギーが増加するとバンドギャップは大きくなります。これは、従来の半導体物理学と非常によく一致するパターンです。
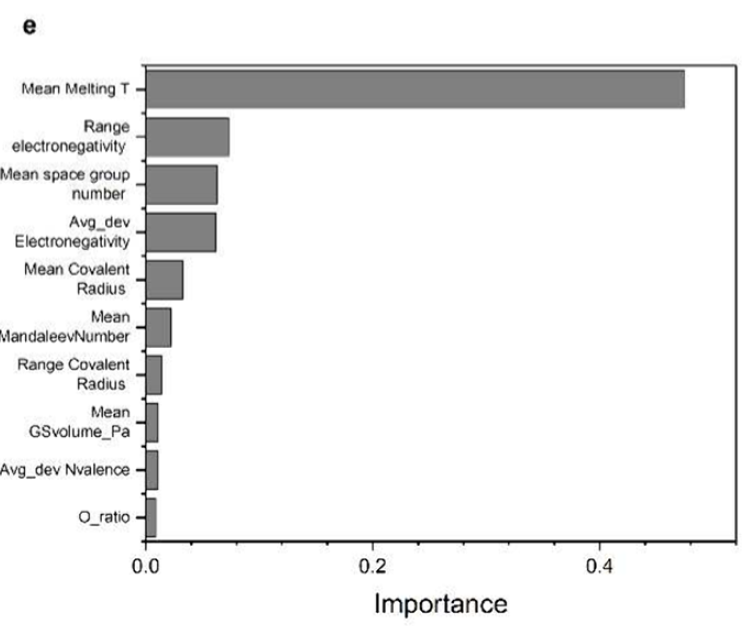
データから実際の化学法則を学習する能力
生成段階では、ベイズ最適化により1,025個のガリウム含有候補成分が提案されたが、そのうちSMACTスクリーニングを通過したのはわずか38個であり、化学的実現可能性の制約が非常に厳しかったことを示している。これらの有効な材料は主に2.0~2.5 eVの範囲に集中しており、この領域ではイオン結合と共有結合の両方の特性を持つ中バンドギャップ半導体を形成しやすいことを意味します。これらの結果は、Ga₂O₃(約4.8 eV)やGa₂S₃(約2.5 eV)などの既知のシステムと非常によく一致しています。
BO 探索プロセスは、既知のガリウム含有化学ファミリー (Ga–O、Ga–N、Ga–As/Sb など) にクラスター化する傾向も示し、これらの領域で Ga₀.₅₁As₀.₁₆N₀.₂₄Sb₀.₁₀、Ga₀.₁₇₁Sb₀.₁₇₅O₀.₃₆₇F₀.₂₈₆ などの新しい中間化学量論を提案します。
バンドギャップの広い材料(>3.0 eV)の場合、アルゴリズムは酸素を多く含む化合物を優先します。これは、強いGa-O結合がバンドギャップを広げるのに役立つためです。一方、バンドギャップの狭い材料(約1.5~2.0 eV)は、通常、酸素を硫黄、セレン、またはリンで置換することによって得られ、より強いp-p相互作用が導入されます。これらのパターンは既存の実験結果と非常に一致しており、モデルがデータから実際の化学規則を「暗黙的に学習」できたことを示しています。
現実世界の「構造と特性の関係」を捉える能力
生成されたガリウム含有組成物が「物理的に実現可能な」材料であることを確認するため、研究チームはParkらが開発したChemelon-dngモデルを用いて、下図に示すような結晶プロトタイプを予測した。
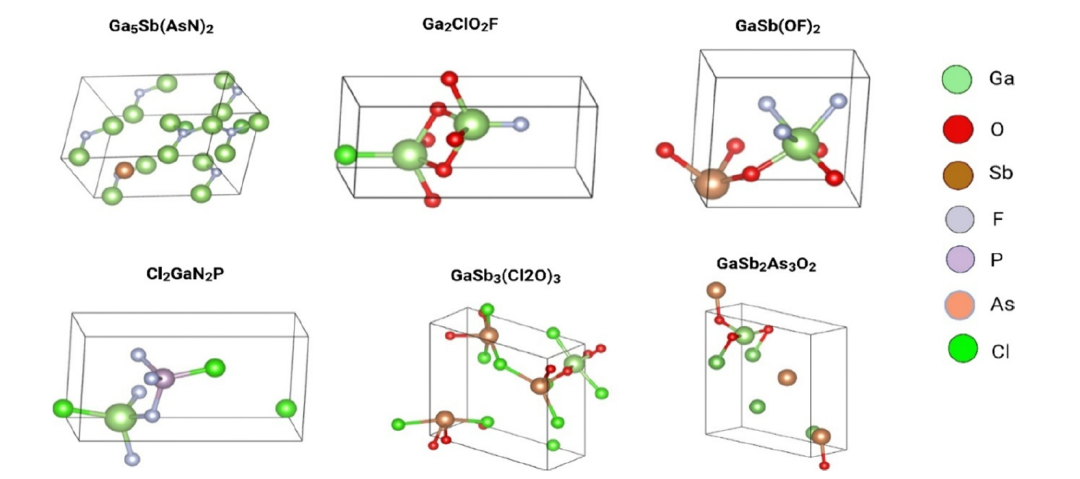
SMACTによって検証された候補成分は、化学的に妥当な配位環境を示し、四面体および八面体のガリウム中心が支配的であり、これはGa₂O₃、GaN、GaSeなどの既知の結晶プロトタイプと非常に一致しています。サロゲートモデルは、経験的に一致する電子構造階層関係、すなわち酸化物:3.5~4.8 eV、カルコゲニド:1.8~2.6 eV、およびA族窒素化合物:約1.2~2.0 eV、つまり酸化物バンドギャップ > カルコゲニドバンドギャップ > A族窒素バンドギャップをうまく再現しました。
この結果は、このベイズ最適化ワークフローは、現実世界の「構造と特性の関係」を効果的に捉えることができるようになった。
注目すべきは、検証された38の有効な成分のいずれも、既存の既知の物質の重複ではなかったことであり、これは生成された結果が「新規性」と「化学的一貫性」の両方を備えていることをさらに証明している。
DFT検証
研究者らはさらにDFT検証を実施した。以下の表は、SMACT検証に合格した10個の構成要素について、「モデル予測バンドギャップ」と「DFT計算バンドギャップ」の比較結果、および対応するバンドギャップの種類をまとめたものである。
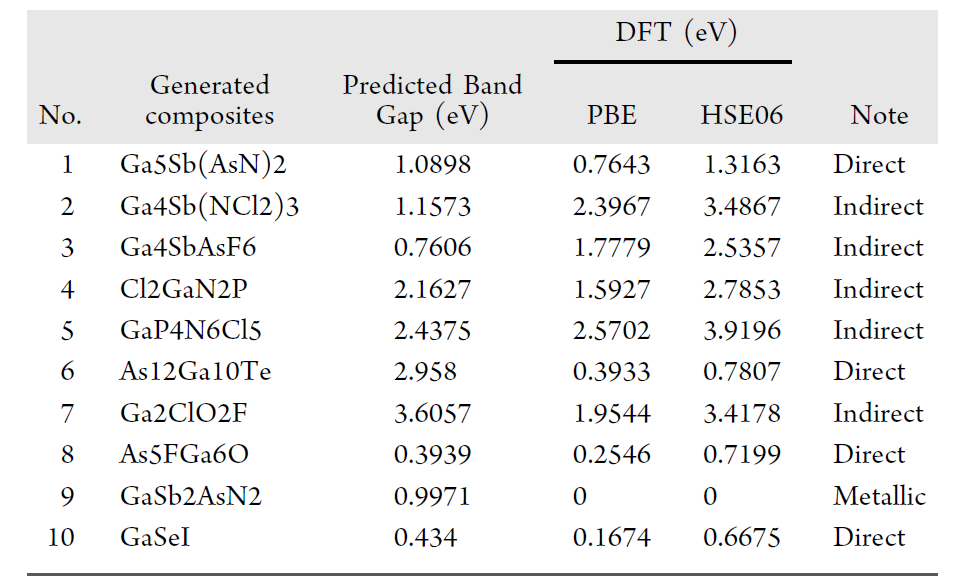
全体として、平均絶対誤差(MAE)は0.890 eV、二乗平均平方根誤差(RMSE)は1.158 eV、中央値絶対誤差は0.784 eVでした。多少の偏りはあるものの、材料探索の初期スクリーニング段階において高い実用性を有しています。さらに重要なことに、検証された材料はすべて既知のデータベースには存在せず、高い新規性を示しています。
結論
総じて、本研究はガリウム含有半導体のための斬新な材料設計パラダイムを実証するものである。それは、機械学習モデリング、ベイズ最適化探索、および化学的制約スクリーニングの相乗効果を通じて、「データ」から「新素材」へと自動的に生成される経路である。
産業界の観点から見ると、このアプローチは太陽光発電材料設計、発光デバイス開発、ワイドバンドギャップ半導体研究において大きな可能性を秘めている。特に、次世代パワーエレクトロニクスおよび光電子デバイスの急速な発展を背景に、バンドギャップ制御可能な材料への需要は急速に高まっており、AIを活用した材料設計手法は、このプロセスを加速させる重要なツールとなることが期待される。
さらに、このフレームワークの重要性はガリウム系に限定されるものではありません。その手法はインジウム、スズ、さらには鉛フリー半導体系にも拡張可能であり、複雑な多成分化合物の合理的な設計のための一般的な道筋を提供します。これは材料科学における新たな段階を示しており、「経験に基づく試行錯誤」から「アルゴリズムに基づく生成」へと移行し、人工知能が化学規則と材料発見を結びつける中心的な架け橋となるでしょう。
参考文献:
https://techxplore.com/news/2026-05-ai-discovery-gen-chips-electronic.html
https://pubs.acs.org/doi/10.1021/acsmaterialslett.5c01482








