Command Palette
Search for a command to run...
86,000個のタンパク質構造データに基づいて、量子力学計算と組み合わせた機械学習手法を使用して、69個の新しい窒素-酸素-硫黄結合を発見しました。

細胞の「工場」において、窒素-酸素-硫黄(NOS)結合は、環境の酸化還元変化に応じて酵素の活性を調節できる可逆的な「スマートスイッチ」のようなものです。 2021年、ドイツのゲオルク・アウグスト大学ゲッティンゲン校の研究チームは、淋菌のトランスアルドラーゼを研究することで、リジンとシステインの間のNOS結合を発見しました。この研究は、単一の病原体と酵素の研究の範囲を超え、学際的なタンパク質科学、医薬品設計、バイオエンジニアリングの重要な基盤を築きます。
しかし、タンパク質構造データの爆発的な増加と科学界によるタンパク質構造における化学結合の継続的な研究により、新たな問題も発生しています。見落とされている他の NOS 結合または化学相互作用はありますか?
上記の考察に基づき、ジョージ・オーガスタス大学のソフィア・バジ氏とシャラレ・サヤド氏は、革新的な計算生物学アルゴリズム「SimplifiedBondfinder」を開発しました。これにより、タンパク質の共有結合の探究における新たな章が開かれます。研究チームは機械学習と量子力学計算を統合して高解像度X線結晶構造データベースを構築し、86,000を超える高解像度X線タンパク質構造を体系的に分析しました。69個の新しいNOS結合が発見されただけでなく、それらにはこれまで観察されたことのないアルギニン(Arg)-システインとグリシン(Gly)-システイン間で形成される新しいNOS結合も含まれていました。
この革新的な発見により、タンパク質化学の範囲が広がり、医薬品の設計とタンパク質工学における標的制御が可能になりました。この研究は NOS 結合に焦点を当てていますが、このアプローチは他のさまざまな化学結合や共有結合修飾の研究にも柔軟に適用できます。構造的に解決可能な翻訳後修飾 (PTM) が含まれます。
研究結果は「タンパク質構造の体系的な再評価によるアルギニン-システインおよびグリシン-システインNOS結合の解明」というタイトルでCommunications Chemistry誌に掲載されました。
研究のハイライト:
* NOS結合はリジン(Lys)とシステインの間にのみ存在するという科学的通説を打ち破り、アルギニン-システインおよびグリシン-システインNOS結合の新しい酸化還元調節機構が革新的な方法で初めて明らかにされました
* 提案された方法は、機械学習、量子力学計算、高解像度X線結晶構造解析データを統合し、この研究分野における体系的な化学結合発見アルゴリズムの欠如という課題を解決し、従来の実験の限界を打ち破り、その後の研究のための信頼性が高く使いやすいツールを提供します。
* 機械学習と人工知能技術により、研究コストは大幅に削減され、研究効率も向上し、タンパク質機能の解明や新たなタンパク質相互作用の特定における機械学習主導技術の好例となっている。

用紙のアドレス:
https://www.nature.com/articles/s42004-025-01535-w
AIフロンティアに関するその他の論文:
https://go.hyper.ai/UuE1o
データセット: 多層的な制限を設けた信頼性の高いデータセットの抽出
SimplifiedBondfinder によって収集されたデータは、3 つの異なるタンパク質データベースから取得されます。これらはPDB、PDB-REDO、BDBです。収集されたデータは、信頼性が高く利用可能なデータセットを選別するために、様々な制約を受けます。その中でも、PDB-REDOデータベース(2024年1月時点)は、PDBの静的構造を改良・最適化し、現代の結晶構造標準に沿うようにしています。オリジナルのPDBエントリと比較して、精度と信頼性が向上しています。下図の左側に示すように:
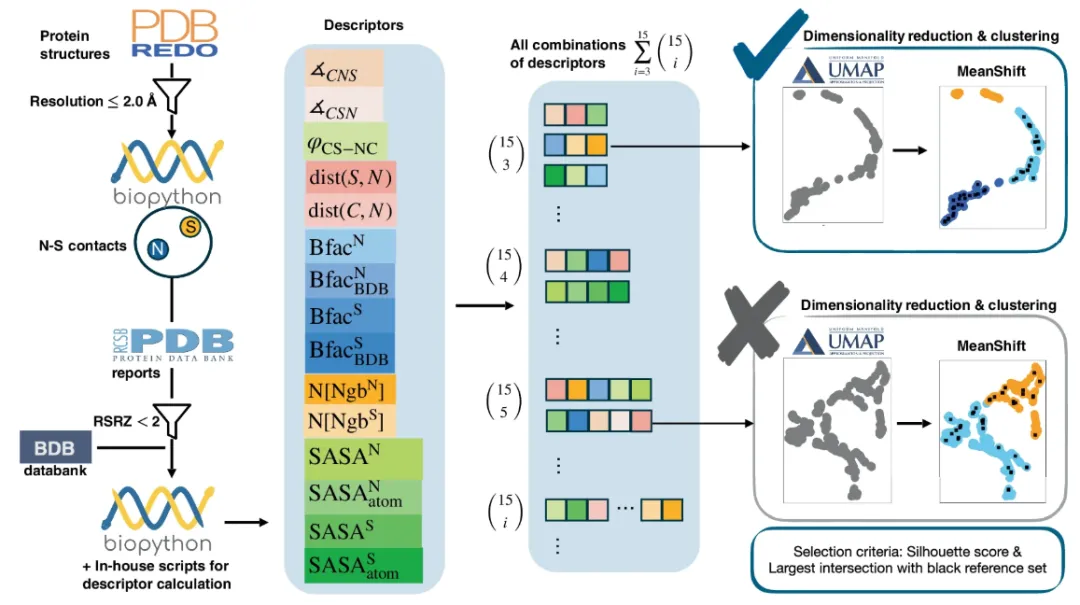
具体的には、研究チームは複数の相互に関連する関数を使用して、当初 170,251 個のタンパク質データが含まれていたデータベースで自動データセット生成を実行しました。まず、Biopython(バージョン1.79)を用いて構造解析(MMCIFParserとPDBParseを使用)を行い、その他の原子および残基特性を計算しました。X線で決定された構造のみを解析した後、研究チームは170,127個のタンパク質データを最適化しました。
その後、研究チームは予測精度をさらに向上させるために、解像度2Å以下のタンパク質構造をさらにスクリーニングし、最終的に実験分析用の86,491個の構造を取得しました。
特定の化学結合を研究するためのデータセットを構築するには、研究チームは、構成原子の種類、残基名、原子間距離、占有率に基づいて基準を確立しました。標準残基中の硫黄(S)原子と窒素(N)原子が関与するNOS結合について、研究チームはSNの原子間距離(dist(S, N))をリジンとシステイン間の原子価相互作用のカットオフ値に相当する3.2Å以下に制限し、位置の不確実性が高い原子を除外するために占有閾値を0.8以上とした。この基準を用いて、25,462個のNS結合が同定された。
対象の原子質量が確実に描写されるように、研究チームはさらに、閾値を 2.0 未満に設定した実空間 R 値 Z スコア (RSRZ) を適用し、実空間のデータとの信頼性の高い一致を識別できるようにしました。データセットはさらに 23,129 件の NS 連絡先に削減されました。これにより、実験対象は主にシステインの 2 種類の相互作用、すなわちシステインの硫黄原子とグリシンの主鎖窒素との相互作用、およびシステインの硫黄原子とアルギニンおよびリジンの側鎖窒素との相互作用に集中できるようになりました。
次、研究チームは、Biopython の NeighborSearch モジュールを使用して構造パラメータを抽出し、各データセット内の各サンプルに対して 15 個の異なる記述子を収集しました。これらには、角度 (∡CSN、∡CNS)、ねじれ角 (φCS-NC)、その他の距離 (dist(C、N)、dist(S、N))、および Bio.PDB.SASA を使用してさらに計算されたターゲット原子と対応する残基の溶媒アクセス可能表面積 (SASA) 値が含まれます。
研究チームは、分析において目標とする原子移動度パラメータを得るために、実験に原子B因子(Bfac)を含めました。これらの値は、RCSB PDBと、B因子が一致するPDBファイルデータベース(BDB)の2つのデータベースから取得されました。
この研究の特定の要件に基づいて、実験では 15 個の記述子のみが選択されたことは言及する価値があります。しかし研究チームは、提案されたアルゴリズムでは処理できる記述子の数に厳密な制限はないと述べた。設計上、任意の数の記述子に対応できるため、ドメイン固有の知識を統合したり、新しい実験的アプローチに適応したりすることができます。
モデルアーキテクチャ:機械学習と量子力学コンピューティングの統合
上記の部分は、提案された方法の重要なステップの最初のステップであり、特定の化学結合の対象データセットを構築し、厳格な基準を適用することです。このセクションでは、提案方法の 2 番目の重要なステップである、機械学習技術を使用してこれらの高次元データを探索することに焦点を当てます。効果的な構造記述子を特定し、共有結合形成の潜在的な場所を予測します。
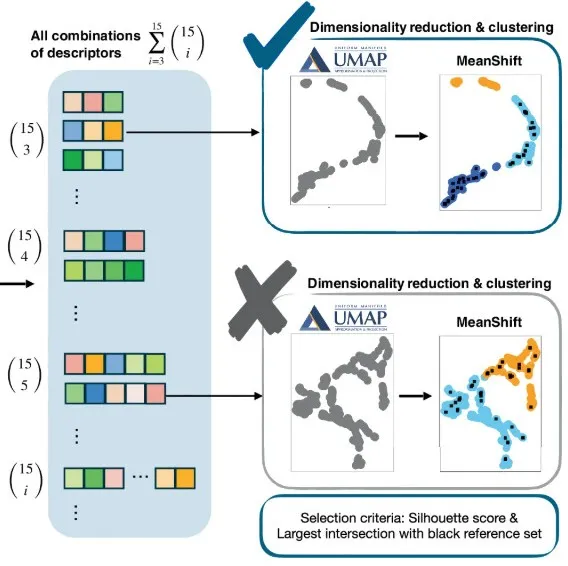
上の写真に示すように。まず、研究チームは、最大埋め込み次元が 3 である教師なしの Uniform Manifold 近似および投影 (UMAP) 次元削減手法を適用しました。次に、すべての可能な記述子セットに対して平均シフト クラスタリングが実行されます。
で、UMAP は、高次元データの固有の位相的および幾何学的特性を最適に保存し、低次元の埋め込みで重要な構造的特徴が保存されることを保証します。これにより、意味のある下流解析が容易になります。UMAPにおける埋め込み次元の選択は、データセットの位相的および幾何学的特性と、元の高次元多様体に依存します。実用的には、2次元または3次元の埋め込みが最も解釈しやすいです。これは、直感的な視覚化とクラスタリング品質の評価が可能になるためです。
本研究では、3次元の埋め込み次元を用いることで、十分に分離され意味のあるクラスターが得られ、その選択が正当化されました。化学結合分析とクラスタリングの結果は、この次元削減手法が本実験で使用したデータセットに最適であることを示しています。必要以上に高い埋め込み次元を選択すると、元の一般的な特徴は維持されますが、解釈可能性は向上せず計算コストが増加します。逆に、最適なレベルよりも低い次元数に削減すると、大量の情報が失われ、クラスターの分離が不十分になります。
続いて、研究チームは、すべての3次元埋め込み座標のシルエットスコアを取得し、それぞれの組み合わせのクラスタリング品質を評価しました。このアルゴリズムは、クラスター、シルエット係数、そして各クラスター内の参照-ターゲット間の接続性を出力します。各候補は、ターゲット原子名、対応する残基名、残基番号、鎖、そしてPDB IDによって識別され、タンパク質内のすべてのターゲット原子を区別します。
最終的な最小の特徴空間を見つけるために、研究チームはシルエット係数の値、各特徴空間によって生成されるクラスターの数、これらのクラスター内の参照ターゲット接続の分布など、いくつかの基準を使用しました。
具体的には、研究チームは、シルエット係数が 0.5 以上の 2 つまたは 3 つの異なるクラスターにデータを効果的に分割できる特徴空間を特定することを目的としました。理想的には、クラスターの1つに参照ターゲット接続が全く含まれず、これは「不可能クラスター」と呼ばれます。実際には、このクラスターに含まれる参照サンプルの最小数は許容範囲です。残りのクラスターには、参照ターゲット接続のすべてまたはほとんどが含まれますが、「可能クラスター」と呼ばれます。
対象となる化学結合を含む可能性のある候補クラスターと不可能な候補クラスターを導入することにより、研究チームは、新しい化学結合を形成する可能性が高いターゲット原子ペアと、そのような結合を形成する可能性が低いターゲット原子ペアを区別するために最適化された特徴空間を特定することができました。これらのケースを確実に区別できる記述子セットが特定されれば、追加の記述子を追加する必要はなくなります。このアプローチは、計算効率と解釈可能性の両方において利点があり、タンパク質構造内の新たな化学結合形成を特定する方法の予測精度を大幅に向上させる可能性があります。
この研究で提案された手法では、機械学習に加えて量子力学計算も統合されています。研究者らは、Lys-NOS-Cys、Gly-NOS-Cys、ARG-NηOS-Cys、およびARG-NεOS-Cys複合体におけるNOS結合の候補となる分子について、構造最適化を行った。構造最適化は、ソフトウェアパッケージGaussian16 – A.03(Gaussian 16、リビジョンC.01)を用いて、B3LYP-D3 (BJ)/def2-TZVPD理論レベルで水中で行われた。最適化された構造について、硫黄原子と窒素原子間の距離(dist (S, N))や角度(∡CSN、∡CNS、∡NOS)など、いくつかの構造パラメータを実験的に計算した。
提案されたクラスタリング法によって予測されたNOS共有結合の存在を検証するために、研究チームは、phenix.refine (バージョン 1.20.1-4487-000) を使用して、4 つの代表的なタンパク質構造を再最適化しました。高解像度結晶構造データとの整合性を確保するため、phenix.molprobityを用いて包括的な構造検証を実施し、幾何学的品質、衝突スコア、立体的相互作用を評価しました。また、phenix.table1を用いて、改良統計、モデル品質指標、立体化学偏差をまとめた完全な検証レポートを作成しました。これらの検証手順により、NOSジャンクションの構造的完全性と電子密度マップとの整合性が確認されました。
実験結果: Arg-NOS-Cys結合とGly-NOS-Cys結合は妥当な共有結合である
提案された方法の有効性を実証するために、研究チームは、記述子の選択、マルチ記述子空間の生化学的意義、クラスター分析、構造および熱力学的検証のための機械学習技術の使用を検討する多数の実験を実施しました。
機械学習を用いた記述子の選択
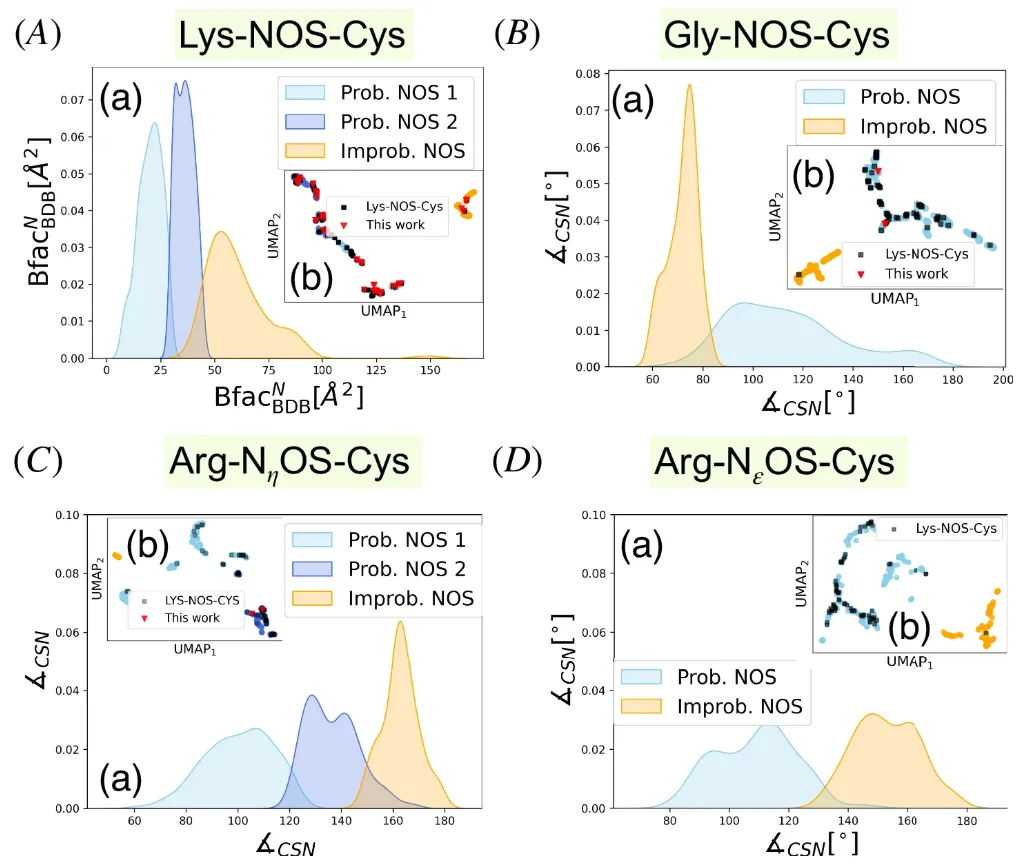
研究チームはまず、Lys-NOS-Cys 結合が存在する可能性のあるデータにこれを適用しました。このデータセットには 527 個のリジン-システイン ペアが含まれており、実験的に検証された NOS 結合も含まれています。重要な記述子は、窒素原子の B 因子 (Bfac(BDB)(N)) とリジン (Ngbᴺ) およびシステイン (Ngbˢ) の Cα 原子の半径 4 Å 以内にある隣接残基の数であることが実験的に決定されました。
研究チームはさらに、313 個のグリシン-システインペアのデータセットに分析を拡大し、下の図に示すように、潜在的な Gly-NOS-Cys 接続を調査しました。
ここで、主要な記述子セットには、硫黄含有残基の B 係数 (BfacBDBS)、硫黄 - 窒素距離 (dist(S,N))、および炭素 - 硫黄 - 窒素角度 (∡CSN) が含まれます。
アルギニンとシステイン残基間のNOS結合形成を予測するための重要な記述子に関しては、アルギニン側鎖には Nη と Nε の 2 種類の窒素原子があり、幾何学的特性と化学的性質が異なります。したがって、Nη (Arg-NηOS-Cys) データセットと Nε (Arg-Nε-Cys) データセットを個別に分析しました。
Arg-NηOS-Cysの場合、選択された記述子は窒素残基の溶媒アクセス可能な表面積(SASAᴺ)、∡CSN、および硫黄(Ngbˢ)と窒素(Ngbᴺ)に隣接する残基に対応します。同様に、240のArg-NεOS-Cysペアのデータセットの場合、主要な記述子はBfacBDBS、SASAˢ、窒素原子の溶媒アクセス可能な表面積、∡CSN、および∡CNSです。
これらの結果は、UMAP 次元削減の視覚化を通じて明確なクラスター分離を示しています。下図に示すように、スカイブルーとロイヤルブルーはNOS結合候補、オレンジは「不可能なクラスタリング」、黒四角は参照データセットを表しています。NOS結合を形成する可能性のあるサンプルの分布が、参照標準点の分布と非常に重なり合っていることが明確にわかります。
多次元記述子空間の生化学的意義
研究チームは、NOS結合と非NOS結合を区別するために重要な主要記述子の生化学的関連性を、アルゴリズム的に最小限の記述子セットを決定することによって調査した。
Bファクターを例に挙げると、異なるクラスターにおけるBファクターの分布パターンは異なります。上記A(a)に示すように、「可能性のあるクラスタリング」と「不可能なクラスタリング」では、Bファクターのモードが異なります。B 係数は原子または領域の柔軟性に関連し、活性部位残基は通常 B 係数が低く、酵素活性に関連していることを示します。しかし研究チームは、B係数が低いことはNOS結合を示している可能性があるが、他の窒素-硫黄相互作用を反映している可能性もあると指摘した。
異なるアミノ酸残基によって形成される NOS 結合の記述子特性に関しては、BfacBDBᴺ が Lys-NOS-Cys の 2 つのクラスターを区別する主な要因です。Gly-NOS-Cys 接続の場合、∠CSN は可能性のある NOS 接続クラスターを区別する主な記述子であり、ほとんどの可能なサンプルで ∠CSN >80° であり、最適化された Gly-NOS-Cys 複合体の ∠CSN 値は約 94° です。∠CSN は、Arg-NεOS-Cys 接続の可能性のある NOS 接続と不可能な NOS 接続を区別するための重要な決定要因です。
クラスター分析
この評価で、研究チームは65個のLys-NOS-Cys結合、2個のGly-NOS-Cys結合(以下の図aおよびb)、および2個のArg-NηOS-Cys結合(以下の図cおよびd)を検出しました。
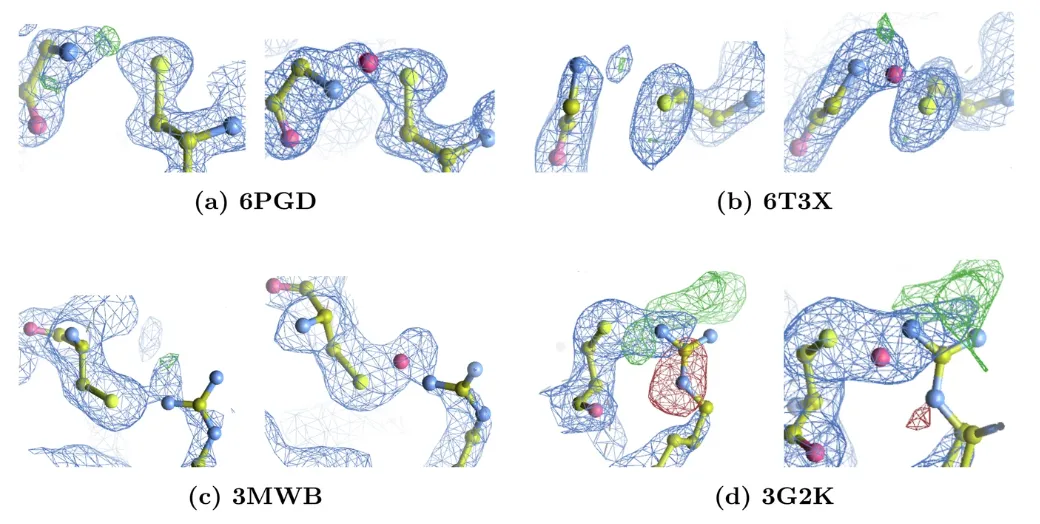
明示的なモデリングと再改良を通じて、研究チームはNOS 結合の導入後、Rwork/Rfree 値は平均 0.5% 改善され、説明のつかない電子密度ピークが大幅に減少しました。3G2Kの場合、元の構造ではアルギニン側鎖付近に負の電子密度ピークが存在しますが、アルギニン配座の再配置後には大幅に減少します。さらに、両モデルともアルギニン側鎖付近に正の差ピークが存在します。これらのピークの振幅が大きく、DMSOが存在することから、現在のモデルではモデル化されていない溶媒分子を表している可能性があります。
構造および熱力学的検証
Arg-NOS-CysとGly-NOS-Cysの関係をさらに確認するために、研究チームは量子力学的形状最適化と4つの代表的なタンパク質複合体(6PGD、6T3X、3MWB、および3G2K)の熱力学的評価を組み合わせ、生体内で起こり得る化学的変動を体系的に説明しました。
構造検証の点では、NOS 結合最適化モデルでは SN 距離は 2.61 ~ 2.70 Å の範囲にあり、これは元の PDB-REDO 構造の 2.63 ~ 2.89 Å 間隔に非常に近いです。橋渡し酸素原子を除去したシミュレーションでは、SN 間隔が 3.36-4.26 Å に大幅に増加しました。これは、実験的に観測された SN 距離の短縮が中間酸素原子の存在と一致していることを示しています。
熱力学的評価の観点から、研究チームはさまざまなプロトン化状態におけるギブス自由エネルギー(ΔG)を計算し、すべてのNOS結合形成プロセスが負であることを示しました。これは、シミュレーション状態において、1つの水素を1つの酸素に置換してNOS結合を形成することが熱力学的に可能であることを示唆している。しかし、ΔGの大きさはプロトン化状態、およびアルギニン由来とグリシン由来の錯体間で大きく異なる。どちらの系においても、中性のグリシンまたはアルギニンは正に帯電した状態よりも優先される。グリシン由来の錯体はわずかに高いΔG値を示す。これらの値は依然として熱力学的に好ましい関係を示唆しているものの、対応するアルギニン錯体よりも系統的に低い発エルゴン性を示す。
これらの構造的結果を総合すると、Arg-NOS-Cys 結合と Gly-NOS-Cys 結合は、単純な非結合接触ではなく、合理的な共有結合であることが示されました。同時に、量子力学的に最適化された形状と結晶系の結晶学データ、および負の形成自由エネルギー間の一致は、これらの接続が関連するタンパク質環境において構造的かつエネルギー的に実現可能であることを強く示唆しています。
機械学習がタンパク質の微視的世界に新たな章を開く
論文でも述べられているように、機械学習と人工知能技術の急速な発展は、生化学における複雑な問題の解決において、従来の生化学的手法よりも優れていることを証明しました。その低い計算コストと高い効率性は、科学研究コミュニティに「生産方法」における大きな革命を促し、機械学習主導の技術がタンパク質機能の解明や新たなタンパク質相互作用の特定においてより大きな可能性を発揮することを促進しました。
偶然にも、カリフォルニア工科大学の Kevin K. Yang 氏らが、Nat. Methods 誌に「タンパク質工学のための機械学習による誘導進化」と題する論文を発表しました。有向進化と機械学習支援有向進化を比較することで、機械学習の優位性を説明します。同時に、酵素触媒効率やシトクロムP450の熱安定性の最適化などの実践事例も挙げており、線形回帰、ガウス過程、ベイズ最適化などのさまざまな機械学習手法についても言及しています。これは、機械学習がタンパク質工学に「データ駆動型インテリジェントナビゲーション」を提供できることを示しています。配列と機能の関係をモデル化することで、指向性進化の効率と成功率を大幅に向上させることができます。
用紙のアドレス:
https://arxiv.org/pdf/1811.10775
さらに、イタリアのボローニャ大学の Rita Casadio 氏らが発表した「タンパク質間の相互作用を予測するための機械学習ソリューション」という論文でも、タンパク質研究における機械学習の探究が詳しく説明されています。タンパク質間分子相互作用 (PPI) における教師なし学習と教師あり学習を含む機械学習手法の応用について紹介します。データ品質、表現、トレーニング アルゴリズム、検証手順における主要な問題が強調されています。
用紙のアドレス:
https://wires.onlinelibrary.wiley.com/doi/full/10.1002/wcms.1618
一般的に、タンパク質のミクロの世界には生命に関わるコードがまだ多く隠されており、機械学習を主な手段として体系的なデータ駆動型の方法は、間違いなくタンパク質のミクロの世界への扉を開く鍵のようなもので、科学研究コミュニティにタンパク質の機能と安定性についてのより深い研究と探究を促し、それによって人類の生命認識の限界を絶えず打ち破っています。








