Command Palette
Search for a command to run...
MIT チームは BoltzGen をオープンソース化し、さまざまな分子タイプにわたるタンパク質バインダーの設計を可能にして、66% ターゲットに対するナノモル親和性を達成しました。

創薬および生体分子工学の分野において、de-novoバインダー設計は自動創薬の中核となる手法です。計算シミュレーションとディープラーニングを用いることで、研究者は特定の標的に結合可能なペプチドまたはタンパク質構造を生成でき、抗体、ナノボディ、環状ペプチドといった新規薬物モダリティの開発が可能になります。
しかし、従来のタンパク質設計戦略は、主に分子動力学シミュレーションや配列最適化アルゴリズムといった物理計算に依存しています。単一のシステムで高い精度を達成できるものの、しかし、計算コストが高く、設計空間が限られており、タンパク質、小分子、RNA などのマルチモーダルターゲットを同時に処理することは困難です。現在の深層生成モデルは生成速度をある程度向上させていますが、一般的に「原子レベル」の構造推論能力を欠き、特定の分子カテゴリーに最適化されているため、汎用性が限られています。さらに、モデル評価はトレーニングセット内の既存の類似複合体に依存することが多く、「未知のターゲット」に対する汎化能力の検証が困難です。制御可能な生成メカニズムと柔軟な構造制約表現が欠如しているため、設計効率と解釈可能性に限界があります。
この問題に対処するために、MITは、ボルツ氏や他の研究機関と共同で、構造予測と複合体設計を統合する「全原子生成モデル」BoltzGenを提案している。このモデルは、従来の個別の残基ラベルを幾何学的な連続表現に置き換えて、単一システムでタンパク質の折り畳みと結合設計の共同トレーニングを実現するだけでなく、分子タイプ全体にわたって制御可能な生成を実現するための柔軟な「設計仕様言語」を構築します。
実験結果によれば、BoltzGen のナノボディおよびタンパク質複合体の設計はすべて、66% に対するナノモル親和性を達成することを目標としています。マルチモーダル生体分子設計において、「単一モデルシステム」が折り畳みと結合性能の同時最適化を実現できることが初めて実証されました。
現在、関連する研究成果は「BoltzGen: ユニバーサルバインダー設計に向けて」というタイトルで公開されています。
GitHub アドレス:
https://github.com/HannesStark/boltzgen
研究のハイライト:
* 単一の全原子生成モデルで構造予測と結合剤設計を統合し、タンパク質の折り畳み、結合部位のモデリング、および原子レベルの精度での配列生成を同時に実行できるため、分子設計の物理的合理性と制御性が大幅に向上します。
* 汎用的な「設計仕様言語」を提案し、タンパク質、ナノ抗体、環状ペプチド、小分子などの異なるシステム間でモデルを柔軟に切り替えることを可能にし、クロスモーダル構造生成と制約制御を実現し、バイオ分子設計分野における生成AIの適用範囲を拡大します。

用紙のアドレス:
https://go.hyper.ai/3sx2K
公式アカウントをフォローし、「BoltzGen」と返信すると、完全なPDFを入手できます。
AIフロンティアに関するその他の論文:
混合データセット: マルチモーダルトレーニング戦略
研究チームは、BoltzGen をトレーニングする際に、マルチレベルのクロスモーダル共同トレーニング フレームワークを採用しました。使用されるデータセットの主なソースには、次の 3 つのカテゴリが含まれます。
* タンパク質データバンク (PDB) からの高品質な実験構造。RNA、DNA、タンパク質小分子などのさまざまな複雑な構造をカバーし、モデルに現実的な化学結合制約と 3 次元幾何学的分布データを提供します。
* AlphaFold2 によって予測および再学習され、実験によって生成された信頼性の高い折り畳みパターンをカバーする AlphaFold データベース (AFDB) からの実験データ。
* Boltz-1 モデルによって生成される複合構造サンプルは、小分子結合や RNA-DNA 相互作用などのマルチモーダル シナリオをカバーしており、さまざまな種類の生体分子にわたるモデルの一般化能力を高めることができます。
モデルが特定の構造タイプに過度に偏ることを防ぐため、研究チームは抗体とTCRのアップサンプリングされたデータセットを排除し、生成された空間の多様性を維持しました。さらに、すべての構造サンプルはトレーニング中にランダムに切り取られ、マルチタスク処理されました。これにより、モデルは各トレーニング反復において、フォールディング予測、複合体設計、構造完成といったタスクをランダムに処理できるようになりました。この統合された多機能学習フレームワークにより、モデルは原子レベルの構造を生成すると同時に、クロスモーダルな理解能力も備えています。
モデルアーキテクチャ:ノイズから構造までの全原子推論
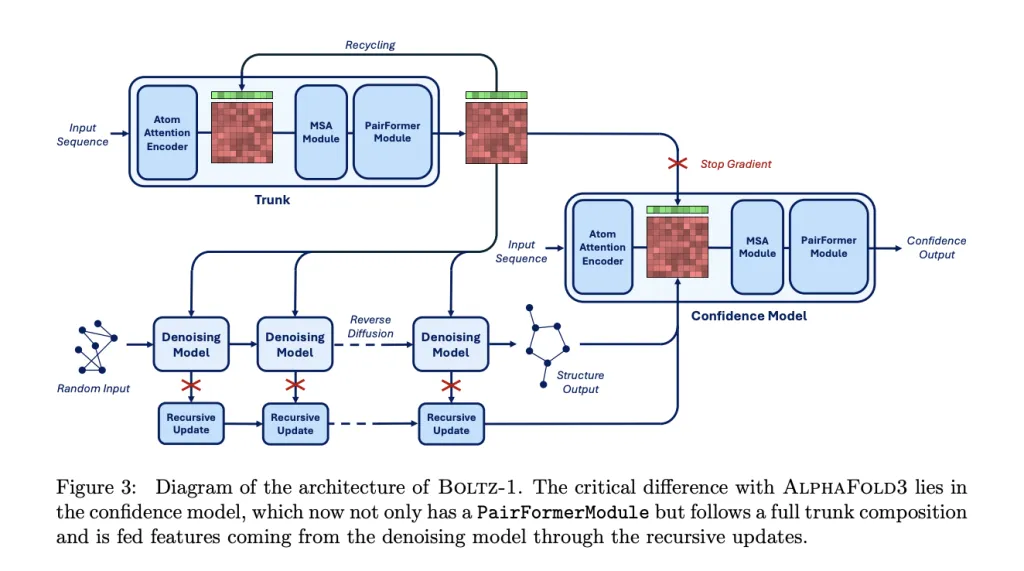
このモデルは、AlphaFold3 および Boltz-2 アーキテクチャの主要コンポーネントを保持し、これに基づいていくつかの改良を加えて、より多くの条件付き入力を導入しています。
下の図に示すように、モデル全体は 2 つの主要な部分に分かれています。より大きなトランク (バックボーン ネットワーク) と拡散モジュール (拡散モジュール)。Trunkモジュールは条件制御のためのトークン表現とペアワイズ表現を生成し、Diffusionモジュールはこれらの表現に基づいて3D構造を生成します。Trunkモジュールは1回のみ実行され、Diffusionモジュールは複数の反復処理を実行して、すべての原子の3D座標から段階的にノイズを除去します。
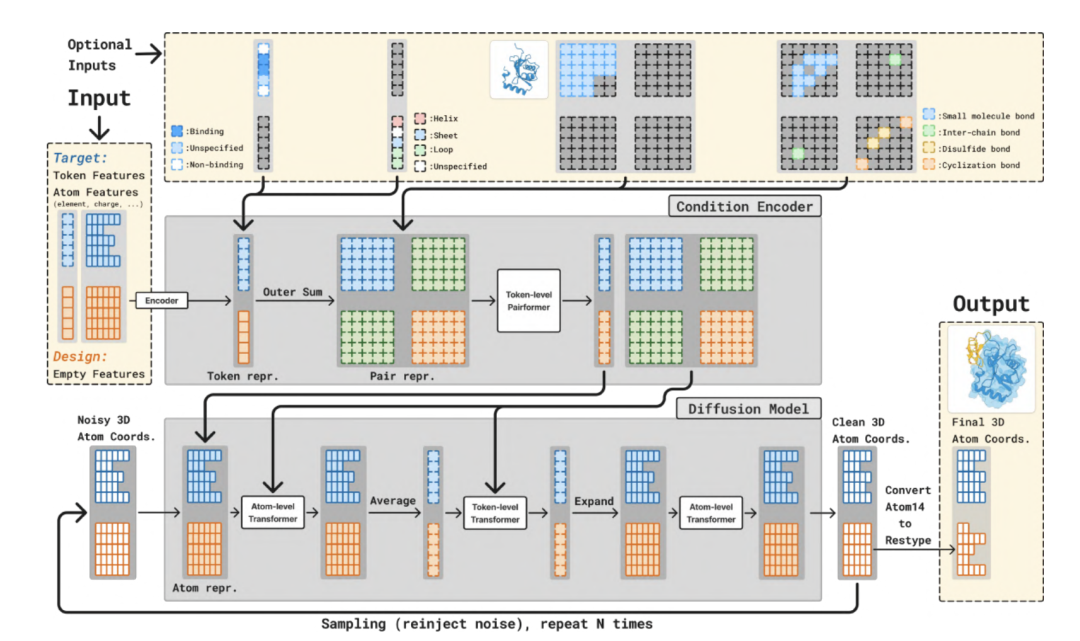
Trunk ステージでは、Boltz-2 の Trunk モジュールに似ており、入力されたタンパク質構造とターゲット情報を解析する役割を担います。 Trunk モジュールはトークン化された分子構造を処理します。メインフレームワークはPairFormerアーキテクチャを採用し、Triangle Attentionを用いて原子間の空間関係を効率的にモデル化します。Geometric Residue Encodingと組み合わせることで、連続空間における残基の種類と原子座標を同時に推定し、離散的なアミノ酸ラベルへの依存を排除します。このメカニズムにより、モデルはデータの記憶のみに頼るのではなく、生成時に構造の物理法則を真に理解することが可能になります。
拡散モジュールの段階では、このモジュールは、ノイズの多い 3D 原子座標を入力として受け取ります。そして、ノイズ除去後の座標を予測します。標準的なTransformerアーキテクチャを採用し、原子レベルとトークンレベルの両方で動作します。BoltzGenは連続空間拡散モデルを用いて、原子座標を徐々に「ノイズ除去」します。ノイズベクトルを予測することでランダムな初期状態を安定なコンフォメーションに変換し、生成プロセス中の分子エネルギー面の制約を維持することで、物理的な衝突や構造崩壊を回避します。
実験結果: 26のターゲットにわたるユニバーサルデザインの検証
実験部分では、BoltzGen モデルのパフォーマンス検証が、タンパク質からペプチド、新規病原体から小分子ターゲットまで複数の次元をカバーし、優れた一般化と制御性を実証しました。
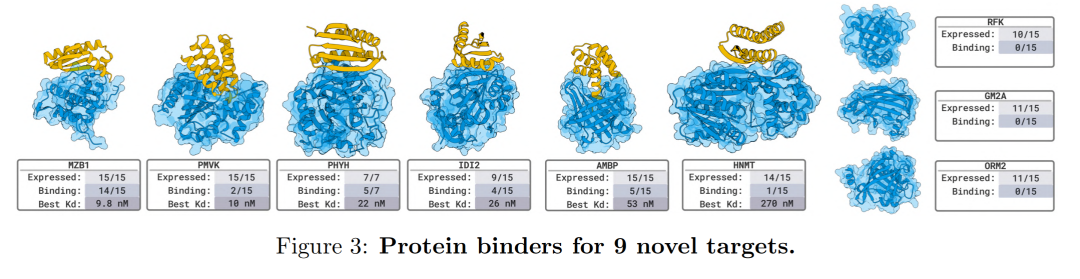
チームは、8 つの独立したウェット ラボ検証プロジェクトで合計 26 個のターゲットをテストしました。結果は、ナノボディ、タンパク質、直鎖状および環状ペプチドなど、様々な結合タイプを対象としています。BoltzGenは、未知の複雑な標的に対して高い成功率を維持しました。トレーニングデータとは全く異なる新規標的を用いた9つの実験において、設計されたタンパク質とナノボディはすべて、66%標的に対してナノモル(nM)レベルの高親和性結合を達成し、モデルの強力な構造推論とクロスモーダル設計能力を実証しました。
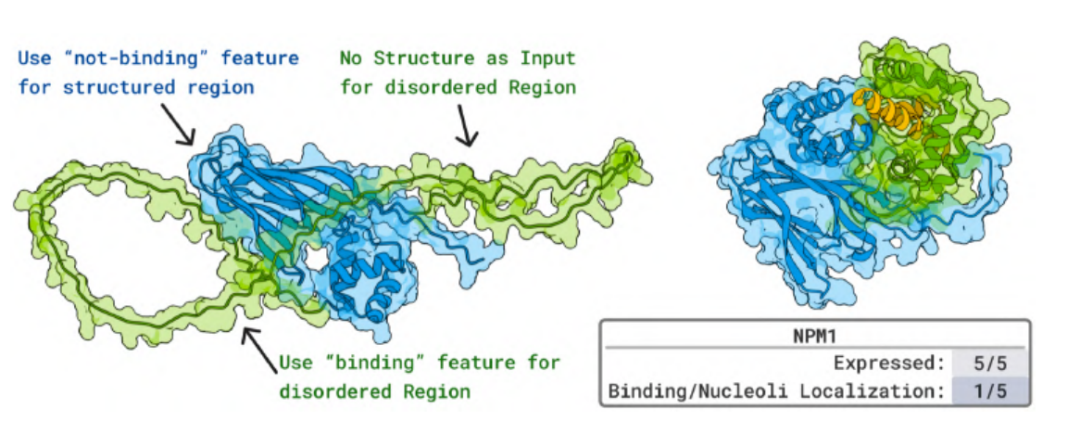
多様な構造を持つ生理活性ペプチドの実験では、BoltzGenによって設計されたタンパク質は、ナノモルからマイクロモル(μM)レベルの親和性で様々な種類のペプチド分子に結合し、それらの抗菌活性または溶血活性を効果的に中和することができます。急性骨髄性白血病に関連する不規則タンパク質NPM1の場合、このモデルによって生成されたペプチドは生細胞内で核小体と共局在することが示され、AIによって設計されたタンパク質が天然に不規則なタンパク質に結合する能力を裏付ける初の生体内証拠となりました。
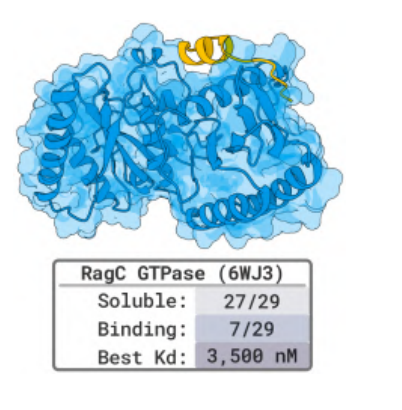
細胞代謝の中核酵素である RagC と RagA:RagC 二量体の設計も注目すべき結果をもたらしました。29 個の候補ペプチドのうち 7 個が RagC に正常に結合し、最高親和性は 3.5 μM に達しました。14 個の環状ジスルフィド結合ペプチド設計は安定した結合を示しました。
BoltzGen はまた、生物医学的に興味深い 2 つの小分子に対して、クロススケール設計機能を実証しました。得られたタンパク質結合剤は50~150µMの範囲で検出可能な結合活性を示し、このモデルが専門的な化学ガイダンスを必要とせずに低分子分子を認識できることを実証しました。さらに、細菌DNAジャイレースGyrAを標的とする抗菌ペプチドの設計において、19%を超える候補配列は細菌の増殖を4倍以上抑制し、一部のペプチドは宿主細胞を直接死滅させました。
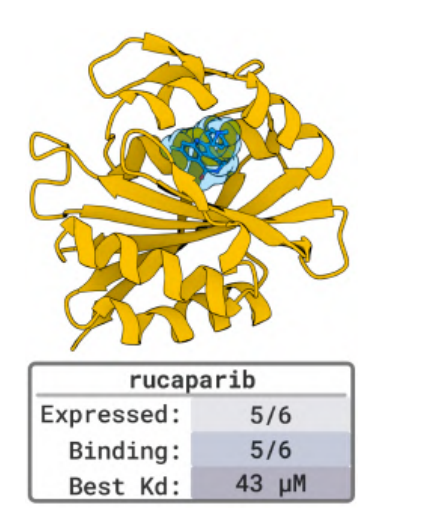
既知の結合構造(PD-L1、TNFα、PDGFRなど)を持つ5つのベンチマークターゲットテストでは、BoltzGen は高いヒット率も達成しました。80% のターゲットにナノモルのバインダーが現れ、その精度が現在の最高モデルと同等であることが確認されました。

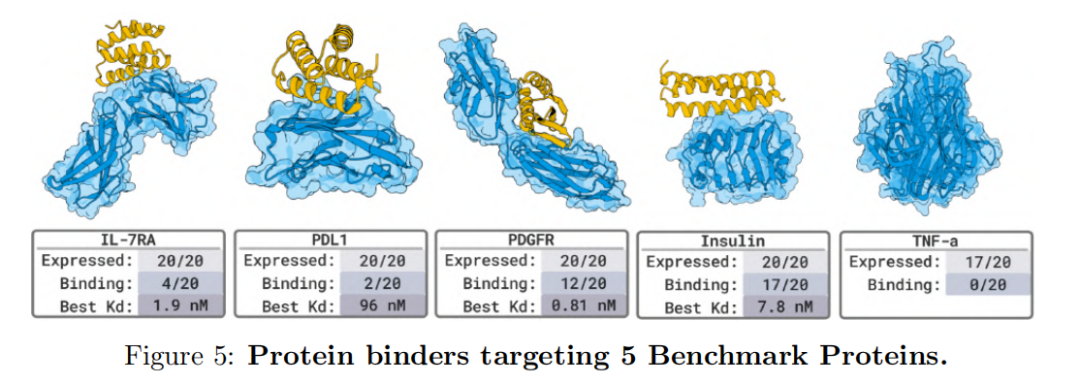
全体として、この一連の実験は、BoltzGenが既知のデータ分布内で高品質な結合構造を再現できるだけでなく、全く未知の生物系における機能設計も実現できることを実証しています。その統合型全原子生成アーキテクチャは、「設計・予測・検証」プロセスを統合し、将来の創薬および生体分子工学のためのオープンで制御可能かつスケーラブルなAIインフラストラクチャを提供します。
予測から生成まで、BoltzシリーズはAI主導の分子設計の展望を一変させる
2024年には、MIT ジャミール クリニックの研究チームは、Boltz-1 モデルを導入しました。世界の医薬品設計業界が構造予測から機能創出へと移行する中、AlphaFoldシリーズのモデルはタンパク質フォールディングの計算可能性を開拓しましたが、AlphaFold3の入手性は限られており、業界が現実世界の医薬品開発シナリオにおいて自由に反復検討を行う能力を制限しています。Boltz-1はこうした状況下で誕生しました。Boltz-1は性能においてAlphaFold3に匹敵するだけでなく、完全にオープンソースで商業的に実現可能なため、分子構造予測を業界のオープンエコシステムへと推進します。
Boltz-1 は、拡散モデルと Transformer アーキテクチャを組み合わせた生成システムを使用します。タンパク質、RNA、DNA、そして低分子複合体の構造を原子レベルで予測できます。柔軟な条件付きインターフェースにより、特定の結合部位や分子の立体配座を正確にモデリングできるため、産業応用が飛躍的に広がります。新規抗体の設計や酵素工学の最適化から低分子リガンドのスクリーニングまで、Boltz-1フレームワーク内でエンドツーエンドの予測を実現できるため、バイオコンピューティングへの参入障壁が大幅に低下します。
2025年には、MIT ジャミール クリニック チームは、Boltz-1 に基づいた Boltz-2 モデルを導入しました。これにより、タンパク質の折り畳み予測の精度が新たな高みに到達し、「構造生物学の GPT-4」として知られています。
Boltz-2は、前身と比較して生成精度と計算効率が大幅に向上しています。また、マルチモーダル条件入力を導入することで、配列情報、実験データ、化学特性を統合し、より洗練された分子設計を可能にします。バイオコンピューティングと創薬の分野が「フルシナリオ生成」へと移行する中で、Boltz-2は、高可用性、スケーラビリティ、そして商業的に実現可能なツールに対する学界と産業界の需要にさらに応えます。
Boltz-2 は、拡散モデルと Transformer アーキテクチャのハイブリッド生成システムを継承し、最適化します。コアとなる Trunk モジュールは、タンパク質または核酸複合体の多段階表現を一度に抽出できます。Diffusion モジュールはこれに基づいて構造を生成し、最適化します。
柔軟な条件付きインターフェースのおかげで、研究者は特定の結合部位、活性ポケット、または低分子リガンドの出力構造を正確に制御することができ、新規抗体設計、酵素触媒の最適化、医薬品リードスクリーニングといった分野におけるモデルの応用可能性を大幅に拡大します。Boltz-2はオープンソースであるため、学界と産業界をまたがる自由な反復処理が可能で、実際の医薬品開発シナリオにおける分子生成計算の応用を加速させます。
現在、BoltzGenは、タンパク質、ナノ抗体、環状ペプチド、小分子などの異なるシステム間でモデルを柔軟に切り替えることを可能にする汎用的な「設計仕様言語」を提案し、クロスモーダル構造生成と制約制御を実現し、バイオ分子設計分野における生成AIの適用範囲をさらに広げています。








