Command Palette
Search for a command to run...
L'Université De Toronto Et d'autres Ont Proposé dnaHNet, Qui Améliore La Vitesse d'inférence De 3 Fois Et Réduit Le Coût De Calcul De l'apprentissage Du Génome De Près De 4 fois.

Le génome est le support de toute l'information génétique d'un organisme, déterminant la fonction cellulaire, le développement individuel et la direction de l'évolution des espèces.La « syntaxe de l'ADN » cachée dans la séquence constitue les règles fondamentales de la vie et représente l'un des problèmes centraux que la biologie moderne doit résoudre de toute urgence.La compréhension de cette grammaire n'est pas seulement liée aux connaissances scientifiques fondamentales, mais elle influe aussi directement sur le développement d'applications clés telles que le diagnostic des maladies, le développement de médicaments et la biologie synthétique.
Ces dernières années, les modèles de base pré-entraînés sur des données de séquences à grande échelle sont progressivement devenus une voie importante pour résoudre ce problème. Grâce à l'amélioration continue de la puissance de calcul, de la taille des données et des paramètres des modèles, ces derniers ont affiché une croissance de leurs performances similaire à la « loi d'échelle ». Des modèles tels que Nucleotide Transformer et Evo ont étendu leur échelle de paramètres à des milliards et ont été entraînés sur des séquences inter-espèces, réalisant des progrès significatifs dans des tâches comme la prédiction des effets de la variation et l'analyse des éléments régulateurs.
Cependant, les séquences d'ADN sont essentiellement des chaînes continues de nucléotides aux contours flous, ce qui constitue une différence fondamentale avec le langage naturel. Les deux principaux paradigmes de modélisation actuellement utilisés sont :La segmentation par mots fixes et la modélisation au niveau d'un seul nucléotide présentent chacune un compromis clair entre puissance expressive et efficacité de calcul :La première méthode risque d'endommager les unités fonctionnelles biologiques, tandis que la seconde engendre des coûts de calcul élevés. Par conséquent, trouver un meilleur équilibre entre faisabilité informatique et fidélité biologique est devenu un enjeu majeur. La segmentation dynamique des mots, en tant que solution potentielle, nécessite encore une exploration systématique.
Dans ce contexte,Le modèle dnaHNet, proposé conjointement par l'Université de Toronto, l'Institut Vector pour l'intelligence artificielle au Canada et l'Institut Arc aux États-Unis, entre autres institutions,Cette approche offre une nouvelle solution pour surmonter les difficultés mentionnées précédemment. Les résultats de recherche associés, intitulés « dnaHNet : un modèle de base hiérarchique et évolutif pour l’apprentissage des séquences génomiques », ont été publiés en prépublication sur arXiv.
Points saillants de la recherche
* L'efficacité de calcul de dnaHNet surpasse celle de StripedHyena2, et sa vitesse d'inférence est plus de 3 fois plus rapide que celle de Transformer.
* Nous proposons des stratégies d'entraînement optimales telles que la planification du taux de compression et l'équilibrage encodeur-décodeur.
* A atteint l'avant-garde dans les tâches sans échantillon, telles que la prédiction des effets de variation et la classification de la nécessité des gènes.
* Il peut apprendre la segmentation des mots biologiques en fonction du contexte et s'adapter aux régions fonctionnelles telles que les codons, les promoteurs et les régions intergéniques.

Adresse du document :
https://arxiv.org/abs/2602.10603
Suivez notre compte WeChat officiel et répondez « dnaHNet » en arrière-plan pour obtenir le PDF complet.
Conception d'ensembles de données génomiques multiniveaux pour l'entraînement et l'évaluation des modèles
Pour faciliter l'entraînement du modèle et l'évaluation du système, cette étude a construit un système de données multicouche.Les données de pré-entraînement proviennent d'un sous-ensemble traité de la base de données de classification du génome (GTDB).Le processus a rigoureusement respecté les procédures de filtrage, de contrôle qualité et d'élimination des redondances du jeu de données OpenGenome du modèle Evo. Les critères de sélection comprenaient des indicateurs clés tels que l'intégrité de l'assemblage, le niveau de contamination et le contenu en gènes marqueurs ; après sélection, un seul génome représentatif a été retenu pour chaque groupe d'espèces.
L'ensemble de données final couvre 85 205 procaryotes et contient 17 648 721 séquences.Le nombre total de nucléotides est d'environ 144 milliards. Toutes les séquences ont été extraites du génome complet et divisées en segments non chevauchants pouvant contenir jusqu'à 8 192 nucléotides.
En matière d'évaluation, les chercheurs ont construit un ensemble de test à partir de trois dimensions complémentaires afin d'examiner de manière exhaustive les capacités du modèle. Premièrement,Au niveau de l'aptitude au codage localDouze ensembles de données expérimentales au niveau des nucléotides, totalisant 21 250 points de données, dérivés d'E. coli K12 dans MaveDB, ont été utilisés pour évaluer la capacité du modèle à caractériser la grammaire de codage locale et le paysage de fitness des protéines.
Deuxièmement,En termes d'évaluation fonctionnelle à l'échelle du génome entierÀ partir de la base de données des gènes essentiels (DEG), des étiquettes d'essentialité binaires ont été construites pour 62 espèces bactériennes. Les séquences et annotations pertinentes ont été obtenues auprès du NCBI, et une identité de séquence supérieure à 99% avec le nom de l'entrée DEG a été utilisée comme critère d'étiquetage des gènes essentiels. Ceci a généré 185 226 points de données, qui ont servi à évaluer la capacité du modèle à intégrer les dépendances à longue portée et le contexte génomique.
enfin,En termes d'interprétabilité structurelle,Prenons l'exemple du génome de Bacillus subtilis : sa séquence est divisée en différentes régions fonctionnelles grâce à l'annotation fonctionnelle. La capacité de modélisation structurale est vérifiée par l'analyse de l'alignement entre les résultats de la segmentation et les structures biologiques réelles.
Modèle dnaHNet : Modèle de frontière autorégressive sans segmentation des mots
dnaHNet est un modèle basé sur le génome qui ne nécessite pas de segmentateur explicite.La clé réside dans l'utilisation d'un mécanisme de « blocs dynamiques » permettant au modèle d'apprendre par lui-même les unités structurelles de la séquence.Cette conception évite la fragmentation des segments fonctionnels biologiques par une segmentation fixe en mots et allège la charge de calcul de la modélisation nucléotide par nucléotide, permettant ainsi un meilleur équilibre entre puissance d'expression et efficacité de calcul.
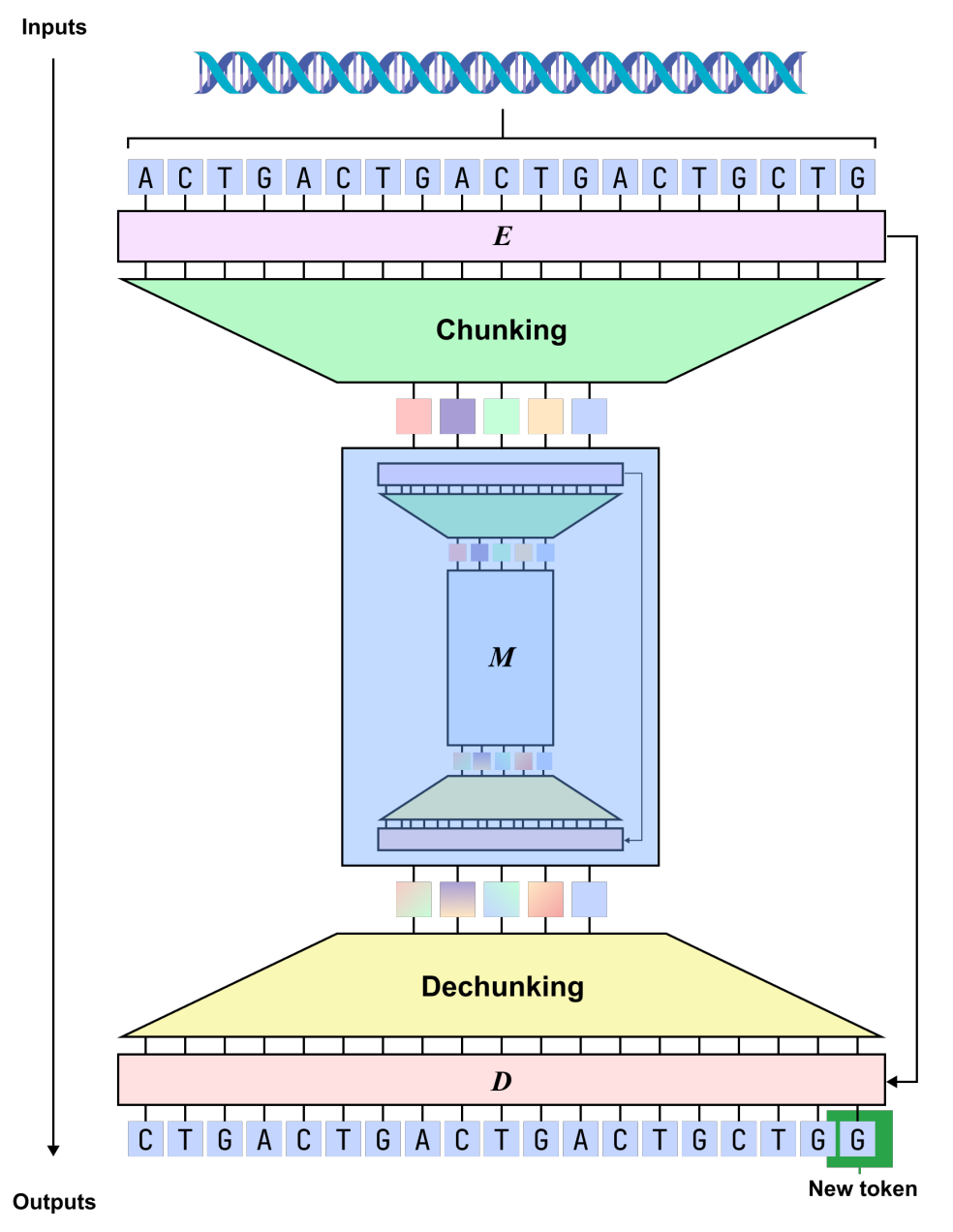
En termes de modélisation,dnaHNet unifie l'apprentissage du génome en une tâche de prédiction de séquence autorégressive, qui prédit le nucléotide suivant en fonction du contexte existant.L'architecture globale adopte une structure hiérarchique, chaque couche étant composée d'un encodeur, d'un réseau dorsal et d'un décodeur : l'encodeur identifie, grâce à un mécanisme de routage, les emplacements de la séquence où l'information change significativement (tels que les limites des codons ou les régions régulatrices) et compresse la séquence en une représentation par blocs implicite ; le réseau dorsal adopte une structure hybride combinant Mamba et Transformer pour modéliser la séquence compressée, en tenant compte à la fois des dépendances à longue portée et des interactions d'informations clés ; le décodeur suréchantillonne ensuite la représentation à la résolution nucléotidique et produit les résultats de la prédiction.
S’appuyant sur ces fondements, dnaHNet a fait l’objet de plusieurs optimisations clés pour les données génomiques. Premièrement, en termes d’allocation des paramètres, environ 301 TP3T de capacité de modèle ont été alloués à l’encodeur et au décodeur afin d’améliorer la capacité à caractériser les structures locales.
Deuxièmement,Intégration d'une conception à compression multicouche en deux étapes :La première étape se concentre sur la capture de motifs à petite échelle (tels que les codons), tandis que la seconde modélise une gamme plus étendue de structures fonctionnelles, assurant ainsi un équilibre entre efficacité de compression et fidélité de l'information. De plus, le processus d'apprentissage intègre une fonction de perte de prédiction autorégressive et des contraintes sur le taux de compression, permettant au modèle de contrôler efficacement les coûts de calcul tout en préservant la précision des prédictions.
Durant la phase d'inférence, le modèle détermine dynamiquement la méthode de segmentation en fonction de la probabilité de frontière, permettant ainsi à la granularité de la modélisation de s'adapter au contexte et de ressembler davantage à la structure réelle du génome.
dnaHNet réduit le coût de calcul de 3,89 fois et surpasse les autres outils multitâches.
Pour évaluer systématiquement les performances de dnaHNet, cette étude l'a comparé à deux modèles de génomes à séquence longue courants : StripedHyena2 et Transformer++.Les expériences couvrent de multiples aspects, notamment les propriétés d'échelle, la prédiction de l'effet de variation à échantillon nul, la prédiction de la nécessité des gènes et la modélisation de la structure biologique.
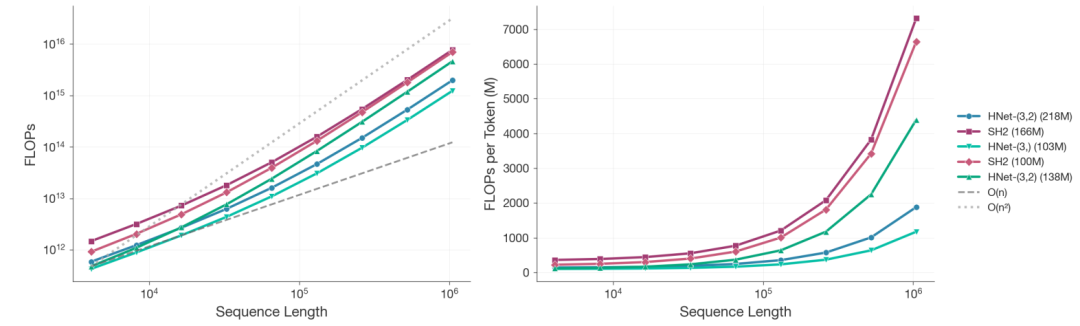
Dans l'analyse d'échelle,Les chercheurs ont entraîné plus de 100 modèles de tailles variables avec un budget de calcul fixe. Lorsque la longueur de la séquence a atteint 10⁶ nucléotides et que la puissance de calcul totale était de 8 × 10¹⁹ FLOPs,Le coût de calcul de dnaHNet avec 218 millions de paramètres est réduit d'environ 3,89 fois par rapport à StripedHyena2 avec 166 millions de paramètres.La structure à deux étages est encore plus efficace que la version à un seul étage.
Les résultats de l'ajustement par loi de puissance basés sur la perplexité montrent que,Pour atteindre le même niveau de performance, StripedHyena2 nécessite environ 3,75 fois la puissance de calcul de dnaHNet.De plus, la configuration optimale des paramètres de données de dnaHNet s'écarte considérablement de la loi d'échelle traditionnelle : à puissance de calcul égale, son nombre de jetons d'entraînement peut atteindre 140 milliards, tandis que le modèle de comparaison n'en compte que 68 milliards et n'a pas encore convergé.
Dans les tâches en aval,dnaHNet surpasse systématiquement les modèles de comparaison tant dans la prédiction de l'effet de la variation des protéines à partir d'un échantillon nul (MaveDB) que dans la prédiction de la nécessité des gènes (DEG).De plus, ses avantages s'accroissent avec l'augmentation de la puissance de calcul. Cela indique que son mécanisme dynamique par blocs et son architecture en couches permettent une intégration plus efficace de la syntaxe de codage locale et des informations contextuelles globales, améliorant ainsi sa capacité à caractériser les fonctions biologiques.
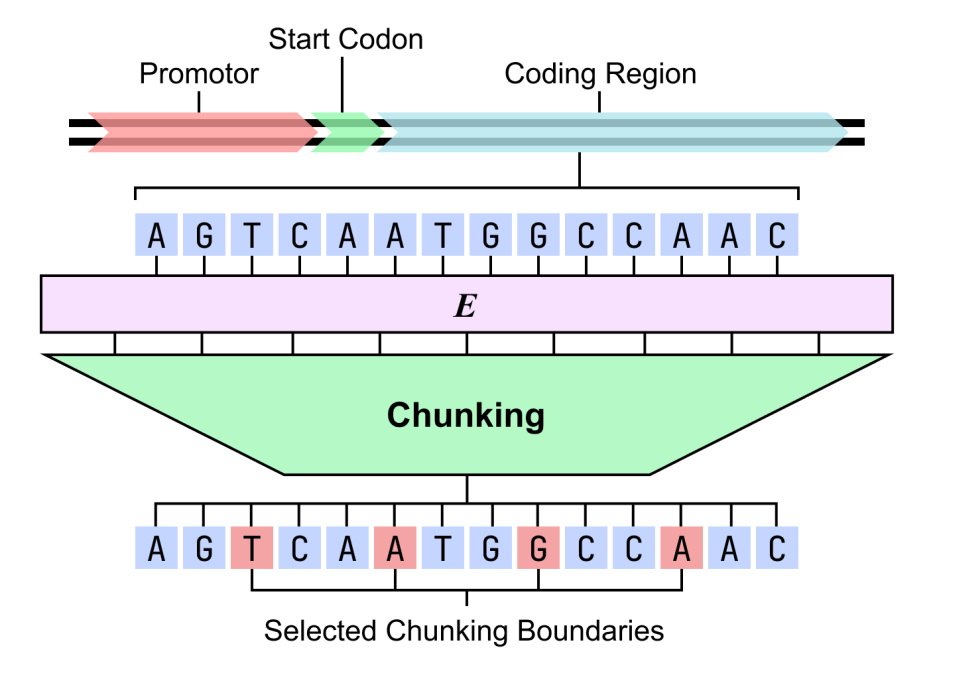
Concernant l'interprétabilité structurale, le génome de Bacillus subtilis a été analysé à l'aide d'un modèle dnaHNet en deux étapes. Les résultats ont montré que le modèle pouvait apprendre spontanément des structures hiérarchiques biologiquement pertinentes : la première étape s'est révélée sensible aux codons et a permis de capturer avec précision les motifs de triplets dans les régions codantes ; la seconde étape s'est davantage concentrée sur les structures fonctionnelles, les promoteurs, les codons d'initiation et les régions intergéniques présentant des probabilités de segmentation significativement plus élevées que les régions codantes.
Ce résultat indique queCe modèle possède non seulement des capacités de prédiction très performantes, mais il peut également reconstituer l'organisation fonctionnelle du génome dans des conditions non supervisées.Cela fournit un chemin de calcul interprétable pour l'analyse de la « grammaire de l'ADN ».
Conclusion
Globalement, dnaHNet ne prédéfinit plus les méthodes de segmentation de séquences, mais laisse le modèle les apprendre automatiquement. Les expériences démontrent que cette modélisation dynamique et hiérarchique améliore non seulement l'efficacité de calcul, mais reflète aussi mieux la structure multi-échelle du génome. À terme, si le modèle parvient à apprendre de manière stable des unités biologiques pertinentes, il est prometteur pour révéler des motifs difficiles à formuler dans le génome, ouvrant ainsi de nouvelles perspectives de recherche en matière de prédiction des variations, de découverte fonctionnelle et de conception synthétique.