Command Palette
Search for a command to run...
Une Équipe Française a Prédit Avec Succès 2,39 Millions De Protéines Antiphages Et a Utilisé Un Modèle d'apprentissage Profond Pour Cartographier l'immunité Antivirale bactérienne.

Dans le monde microscopique, la « course aux armements » entre bactéries et bactériophages est constante. Les bactériophages sont généralement dix fois plus nombreux que les bactéries, ces dernières leur servant d'hôtes pour leur reproduction. Parallèlement, les bactéries ont développé, au cours d'une longue évolution, des systèmes de défense antiviraux extrêmement diversifiés. À ce jour, plus de 250 systèmes antiphages ont été validés expérimentalement, englobant divers mécanismes tels que les systèmes de restriction-modification et les systèmes CRISPR-Cas, et de nouveaux systèmes sont régulièrement découverts. Ce phénomène suggère que la complexité et la diversité des systèmes de défense bactériens pourraient largement dépasser nos connaissances actuelles.Limités par les méthodes expérimentales traditionnelles et les techniques informatiques, un grand nombre de mécanismes anti-phages potentiels restent cachés dans le génome bactérien et n'ont pas encore été explorés de manière systématique.
Les recherches existantes ont mis en évidence certaines caractéristiques communes aux systèmes antiphages connus, tant au niveau de la séquence protéique que de l'organisation du génome, telles que la récurrence de domaines caractéristiques et leur forte concentration dans les « îlots de défense » ou régions préphages. Ces observations suggèrent que :Si ces schémas communs peuvent être identifiés et utilisés, il sera peut-être possible de découvrir systématiquement des systèmes antiphages inconnus à l'échelle du génome entier.
S’appuyant sur cette approche, des chercheurs de l’Institut Pasteur en France ont développé et optimisé trois modèles d’apprentissage profond complémentaires pour la prédiction à grande échelle de la résistance aux phages. Le modèle ALBERT_DF repose exclusivement sur le contexte génomique local pour l’inférence ; ESM_DF utilise un modèle de langage protéique pour analyser les séquences d’acides aminés ; et GeneCLR_DF intègre les informations de séquence au contexte génomique. Dans un test de référence unifié,GeneCLR_DF a obtenu les meilleurs résultats, atteignant une précision de 991 TP3T et un rappel de 921 TP3T.
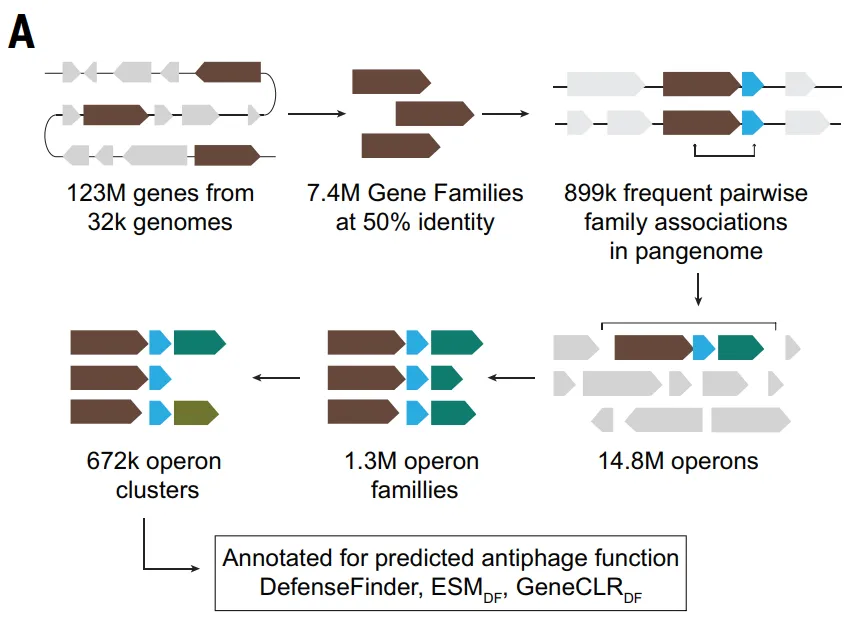
À partir de ce modèle de haute précision, l'étude a ensuite réalisé des prédictions à l'échelle du génome entier concernant les systèmes antiphages. Les résultats ont montré que, parmi plus de 32 000 génomes bactériens, environ 1,51 gène TP3T par génome bactérien typique sont impliqués dans la défense antivirale ; plus important encore, plus de 851 gènes TP3T, appartenant à une famille de protéines prédite comme étant liée à la défense, n'avaient jamais été associés auparavant à la fonction immunitaire. En définitive,Le modèle a prédit environ 2,39 millions de protéines antiphages, dont un grand nombre appartiennent à des systèmes de défense monogéniques, et a défini environ 23 000 familles d'opérons sur la base des relations de cooccurrence des gènes.La grande majorité de ces bactéries étaient jusqu'alors inconnues pour leur rôle dans la défense antivirale. L'ensemble de ces résultats dresse un tableau systématique de l'immunité antivirale bactérienne, révélant son ampleur et sa diversité, bien au-delà des connaissances actuelles.
Les résultats de cette recherche, intitulée « Les modèles de langage protéique et génomique révèlent la diversité inexplorée de l'immunité bactérienne », ont été publiés dans la revue Science.
Points saillants de la recherche :
* Au total, 2,39 millions de protéines antiphages ont été prédites, dont 85% n'avaient jamais été associées à la fonction immunitaire auparavant ;
* Dans un génome bactérien typique, environ 1,51 gènes TP3T sont spécifiquement responsables de la défense antivirale.
* Environ 23 000 sous-familles de manipulateurs ont été prédites, dont la grande majorité ont été découvertes pour la première fois ;
* Un grand nombre de protéines de défense prédites existent sous la forme de systèmes monogéniques, remettant en question l'idée reçue selon laquelle les fonctions de défense sont généralement assurées par la collaboration de plusieurs gènes.

Adresse du document :
https://www.science.org/doi/10.1126/science.adv8275
Suivez notre compte WeChat officiel et répondez « GeneCLR » en arrière-plan pour obtenir le PDF complet.
Ensemble de données : basé sur 123 millions de protéines et 32 000 génomes
Cette étude a d'abord utilisé les outils DefenseFinder et PadLoc,Une analyse systématique de 32 798 génomes bactériens complets dans la base de données RefSeq a été réalisée pour caractériser quantitativement les systèmes antiphages connus.Parmi les quelque 123 millions de protéines, DefenseFinder v1.3 en a identifié 521 360, représentant 0,41 TP3T, qui appartiennent aux composants du système antiphage, tandis que PadLoc en a identifié 805 357, représentant 0,651 TP3T.
Il convient de noter que de nombreux systèmes de défense ont été initialement découverts grâce à des associations génomiques avec des systèmes connus. Ces associations peuvent être quantifiées au niveau de la famille de protéines à l'aide d'un « score de défense », qui mesure la fréquence à laquelle une famille de protéines particulière coexiste avec des protéines de défense connues dans le génome.
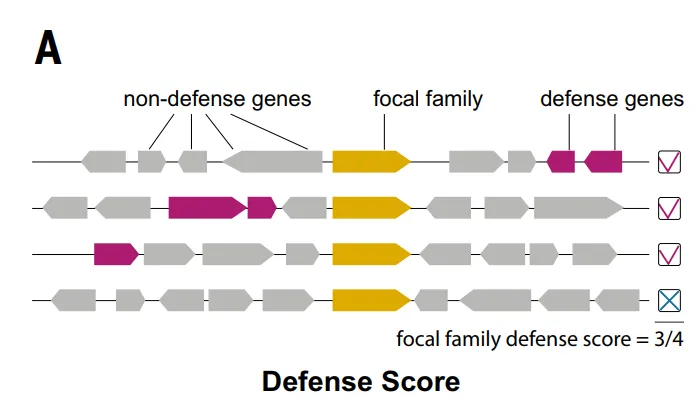
D’après la méthode de notation défensive, comme illustré dans la figure ci-dessous.Les chercheurs ont identifié un total de 37 959 familles de protéines (4,61 % de TP3T) comme familles d'antiphages candidates.Par la suite, l'étude a éliminé 7 799 familles, telles que les intégrases, qui étaient associées à des fonctions biologiques essentielles ou à des éléments génétiques mobiles, aboutissant finalement à 30 160 familles candidates sélectionnées (représentant 3,71 TP3T).
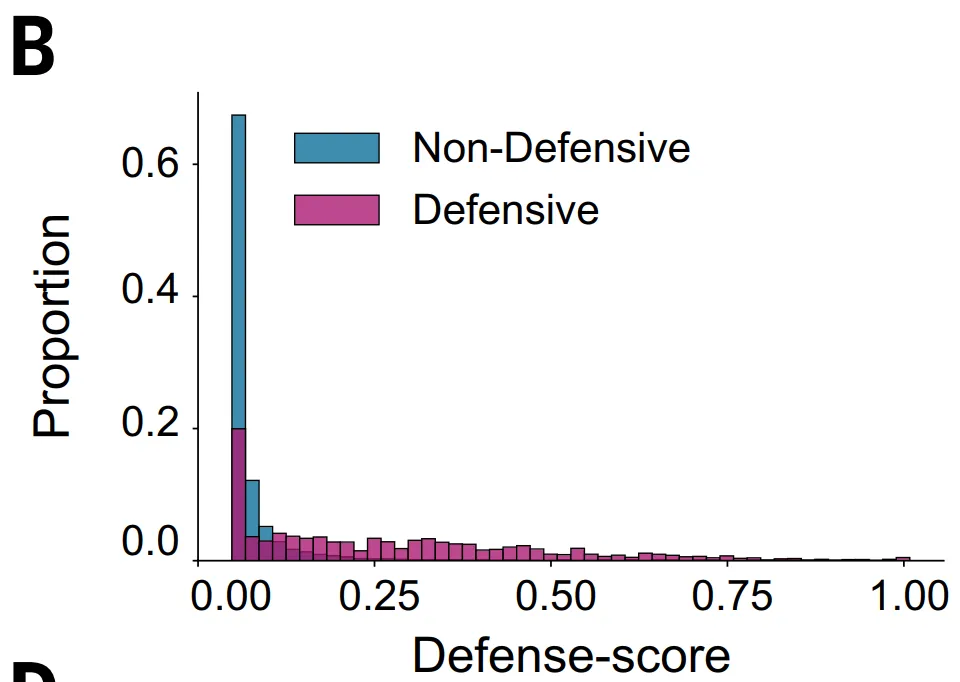
Cependant, cette méthode présente des limites évidentes :Premièrement,Cela ne s’applique qu’aux familles de protéines contenant plus de cinq séquences homologues, excluant ainsi les protéines d’environ 23% ;Deuxièmement,Certains systèmes antiphages ne sont pas situés dans les îlots de défense typiques, et même s'ils ont des fonctions défensives, leurs scores de défense peuvent être faibles, ce qui les fait passer inaperçus.
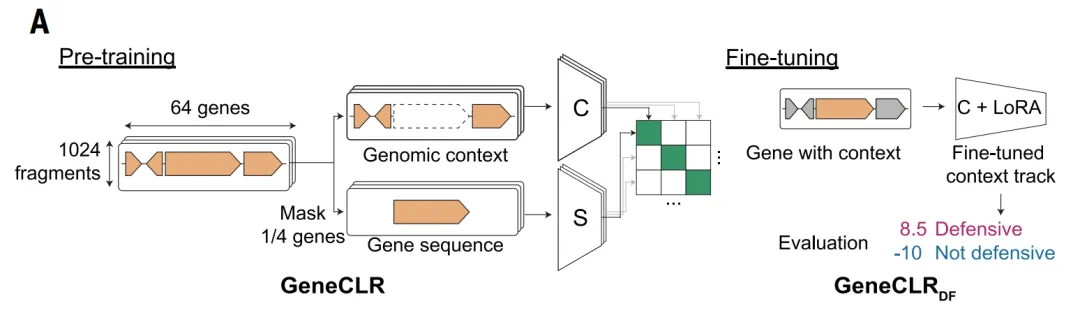
Pour surmonter les limitations susmentionnées et saisir plus complètement les signaux génomiques liés à la défense,L'étude a ensuite permis de constituer un ensemble de données adapté à l'apprentissage profond.Dans le cadre du modèle ALBERT_DF, l'étude a modélisé le génome bactérien de manière « linguistique » : en traitant chaque famille de protéines comme un « mot » et les segments de gènes adjacents comme une « phrase ».
Parce que l'ensemble de données complet contient plus de 8 millions de familles de protéines différentes, dépassant largement la taille du vocabulaire des modèles de langage traditionnels,L'étude a limité le champ d'étude à l'embranchement des Actinobactéries, en constituant un ensemble de données contenant 10 796 génomes.Les gènes ont été regroupés en 4,2 millions de familles de protéines, tandis que le vocabulaire a été limité aux 524 288 familles les plus courantes, couvrant ainsi environ 891 protéines TP3T.
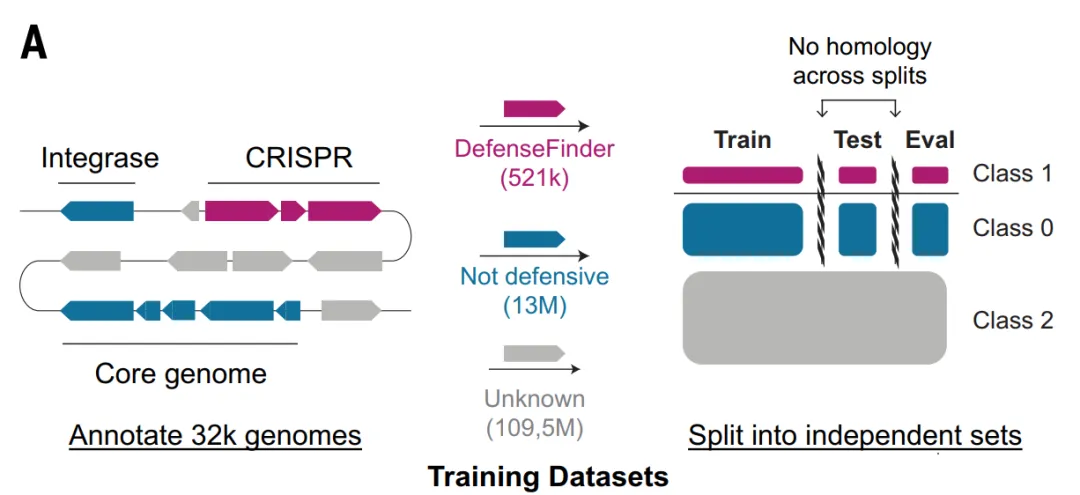
Pour les modèles ESM_DF et GeneCLR_DF, l'étude a construit l'ensemble de données Gembase_DF : comme indiqué dans la figure ci-dessous, 521 360 protéines antiphages marquées avec DefenseFinder ont été utilisées comme échantillons positifs, 116 millions de gènes centraux hautement conservés présents dans plus de 99% et 14 millions de gènes d'éléments génétiques mobiles non liés à la défense ont été utilisés comme échantillons négatifs, et les protéines restantes ont été conservées comme candidats non marqués.
Pour éviter les fuites d'informations entre l'entraînement, la validation et les tests, l'étude a regroupé toutes les protéines du même système de défense dans le même repli de données et a utilisé MMseqs2 pour supprimer l'homologie résiduelle entre les replis de données, garantissant ainsi la rigueur de l'évaluation du modèle.
Architecture du modèle : Un modèle d’apprentissage profond à trois couches qui progresse étape par étape.
Pour surmonter les limites des méthodes traditionnelles de « score de défense », l'équipe de recherche a construit un cadre d'apprentissage profond complémentaire et progressif, ciblant trois objectifs : la découverte de systèmes inconnus, l'exploration à l'échelle du pan-génome et la prédiction intégrée de haute précision.Plus précisément, cela inclut ALBERT_DF basé sur le contexte génomique, ESM_DF basé sur la séquence protéique et GeneCLR_DF qui intègre les informations de séquence et de contexte.
Parmi eux, ALBERT_DF se concentre sur l'apprentissage des signaux fonctionnels à partir des « relations de voisinage » des gènes et a la capacité de découvrir de nouveaux systèmes de défense ; ESM_DF utilise directement la modélisation de la séquence d'acides aminés et possède une bonne capacité de généralisation inter-séquences ; tandis que GeneCLR_DF intègre les deux types d'information dans un cadre unifié et atteint un meilleur équilibre entre la précision de la reconnaissance et la couverture de la prédiction.
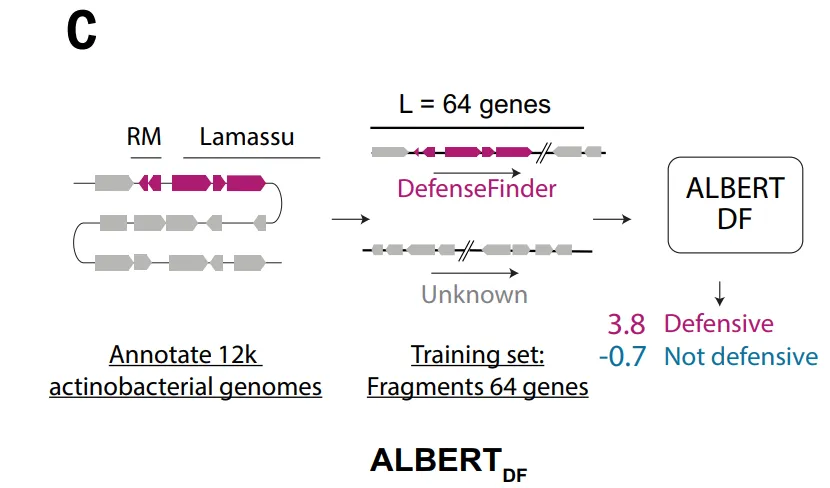
Le modèle ALBERT_DF repose sur une observation clé : les systèmes antiphages ont tendance à être regroupés dans l’ensemble du génome, avec des schémas d’organisation stables existant au sein et entre les gènes voisins. Sur la base de cette caractéristique,Cette étude introduit l'architecture ALBERT issue du traitement automatique du langage naturel dans la modélisation du génome.En traitant les familles de protéines comme des « mots » et les séquences de gènes comme des « structures syntaxiques », nous apprenons le contexte local en prédisant les gènes masqués.
Contrairement aux méthodes traditionnelles basées sur la similarité des séquences, cette approche de modélisation exploite directement l'information d'organisation génomique, offrant ainsi un potentiel accru pour l'identification de nouveaux mécanismes de défense sans homologie avec les systèmes connus. Cependant, du fait de son recours à des représentations « lexicales » discrétisées, ce type de méthode présente des limitations intrinsèques lorsqu'il est appliqué à d'autres espèces.
Le modèle ESM_DF, quant à lui, adopte une approche différente, agissant directement sur la séquence d'acides aminés de la protéine.Ce modèle apprend les covariations entre les résidus et les relations de séquence à longue portée grâce à un pré-entraînement à grande échelle.Cela permet d'extraire des signaux fonctionnels sans recourir à des caractéristiques artificielles. Après un paramétrage précis, ESM_DF peut évaluer n'importe quelle protéine afin de déterminer si elle participe à la défense anti-phages. Cette approche améliore considérablement l'applicabilité de la méthode, lui permettant de fonctionner à l'échelle du pangénome. Cependant, la capacité de discrimination d'ESM_DF reste dépendante, dans une certaine mesure, de la similarité de séquence ; elle est donc plus performante pour identifier des variants éloignés de systèmes de défense connus, et sa capacité à identifier de nouveaux domaines dépourvus d'homologie est relativement limitée.
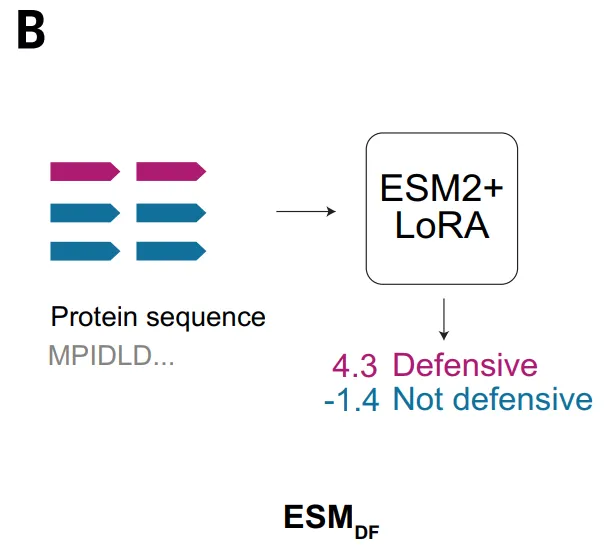
Sur cette base, le modèle GeneCLR_DF a été proposé pour intégrer les informations relatives à la séquence et au contexte génomique.Ce modèle utilise un cadre d'apprentissage contrastif, apprenant simultanément deux représentations pour chaque gène :Une représentation provient de la séquence protéique, l'autre de son environnement génomique. L'entraînement du modèle permet de déterminer si ces deux représentations correspondent au même gène, alignant ainsi les deux types d'information dans l'espace de représentation.
Cette conception offre un avantage clé : lorsque certains gènes ne présentent pas d’homologie au niveau de la séquence, leur contexte génomique typique peut néanmoins fournir des indices d’identification ; inversement, lorsque l’information contextuelle est atypique, les caractéristiques de la séquence peuvent néanmoins permettre la discrimination. Grâce à ce mécanisme complémentaire,GeneCLR offre un équilibre entre la capacité à découvrir de nouveaux systèmes et l'évolutivité nécessaire aux applications à grande échelle pour les prédictions ultérieures.
Globalement, ces trois types de modèles forment une voie technique claire : de l’apprentissage de motifs locaux contextuels à la généralisation globale basée sur les séquences, puis à la modélisation unifiée d’informations multi-sources. Cette conception hiérarchique permet non seulement de s’affranchir des limitations d’une méthode unique, mais aussi de disposer d’un cadre technique plus universel pour explorer systématiquement les mécanismes anti-phages encore inconnus.
Atteignez une précision de 991 TP3T et un rappel de 921 TP3T.
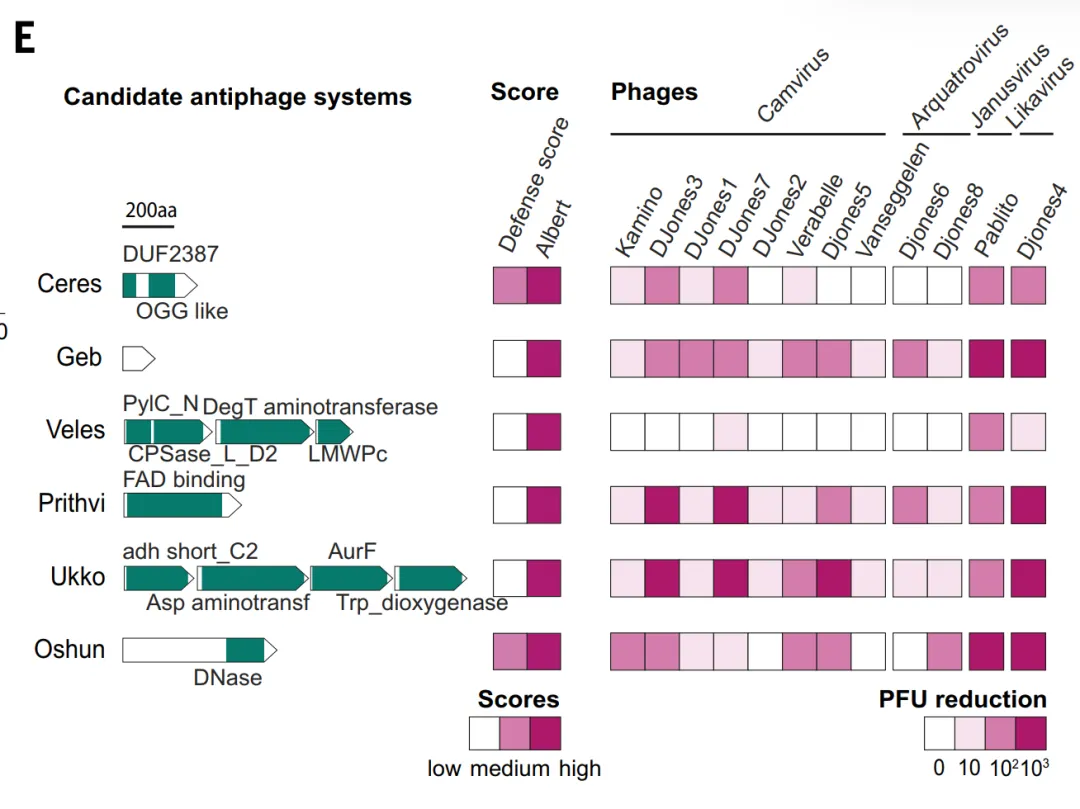
Dans la validation expérimentale, l'étude a d'abord évalué le pouvoir prédictif d'ALBERT_DF.Le modèle a prédit un total de 1 930 familles de protéines antiphages candidates, dont environ 331 TP3T chevauchaient les résultats de la méthode de score de défense.Les chercheurs ont ensuite sélectionné 10 systèmes candidats dépourvus de score de défense et d'homologie connue, les ont exprimés dans *Streptomyces whiteus* et les ont testés avec 12 phages. Six de ces systèmes ont présenté une protection robuste, réduisant les unités formant des plages de lyse de plus de 100 fois. Ces systèmes (tels que Ceres et Geb) contiennent des enzymes métaboliques et de petites protéines aux fonctions inconnues, dépassant le cadre des domaines de défense classiques. Ceci démontre que les méthodes basées sur le contexte génomique peuvent découvrir de nouveaux mécanismes de défense difficiles à identifier par les méthodes traditionnelles.
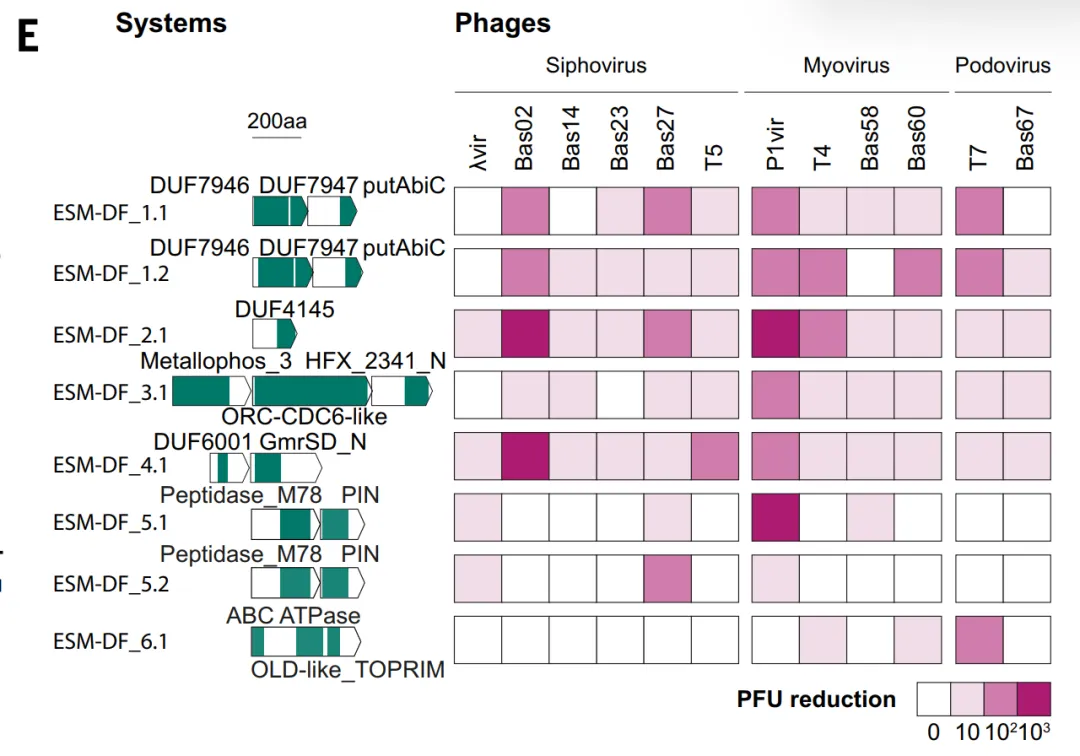
Dans le cadre de la validation d'ESM_DF, l'étude a testé un groupe de candidats prometteurs chez *E. coli*, parmi lesquels six systèmes ont démontré des capacités antiphages, notamment ESM_DF, résistant à plusieurs types de bactériophages. Ces systèmes incluaient des variants de domaines de défense connus ainsi que des domaines non associés auparavant à une fonction antiphage, tels que DUF7946.Cela indique que l'ESM ne se base pas uniquement sur l'homologie de séquence, mais peut également identifier un plus large éventail de caractéristiques fonctionnelles, mais qu'il reste globalement une extension des systèmes connus.
GeneCLR_DF a obtenu les meilleurs résultats lors de l'évaluation du système. Sur l'ensemble de test,Ses scores de prédiction permettent de distinguer clairement les protéines défensives des protéines non défensives.Dans l'analyse évolutive, il a systématiquement attribué des scores élevés à des branches de défense clés telles que les rétrotranscripteurs, CBASS et Thoeris, tandis que ESM-650M_DF ne pouvait les identifier que partiellement.
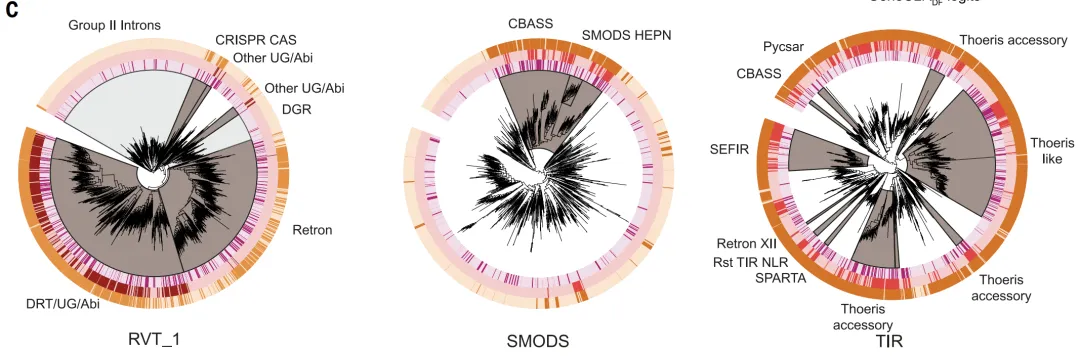
Dans différents contextes génomiques (îlots de défense, intégrons, régions préphages),GeneCLR_DF peut localiser avec précision le module de défense.Les résultats quantitatifs ont montré qu'à un seuil de −0,74, GeneCLR_DF atteignait une précision de 991 TP3T et un rappel de 92,41 TP3T ; à précision égale, ESM_DF n'a rappelé que 581 TP3T. Avec un taux de faux positifs de 11 TP3T, GeneCLR_DF a identifié 941 TP3T dans des familles de défense connues, un nombre significativement supérieur à celui d'ESM-650MDF (351 TP3T) et de la méthode de fractionnement de défense (51 TP3T), et n'a identifié que 561 familles de TP3T ; il a également récupéré 751 TP3T parmi les 110 systèmes nouvellement ajoutés. Sur les 615 672 familles de protéines candidates, 931 TP3T ont été détectées uniquement par GeneCLR_DF.
Au niveau des opérons, une analyse plus poussée basée sur le regroupement colinéaire a révélé qu'un grand nombre de structures de défense demeurent inconnues : la famille de protéines prédite de 85% n'a été identifiée que par ESM_DF et GeneCLR_DF, tandis que la famille d'opérons de 45% et le groupe d'opérons de 52.7% étaient auparavant dépourvus d'annotations fonctionnelles. L'analyse évolutive a également révélé que…La proportion médiane de gènes de défense dans le génome bactérien est passée de 0,46% à 1,53%.De plus, un grand nombre de systèmes sont enrichis en éléments génétiques mobiles, avec 23.5% situé à l'intérieur de la limite MGE et des éléments satellites de 47.1% qui devraient coder des capacités de défense.
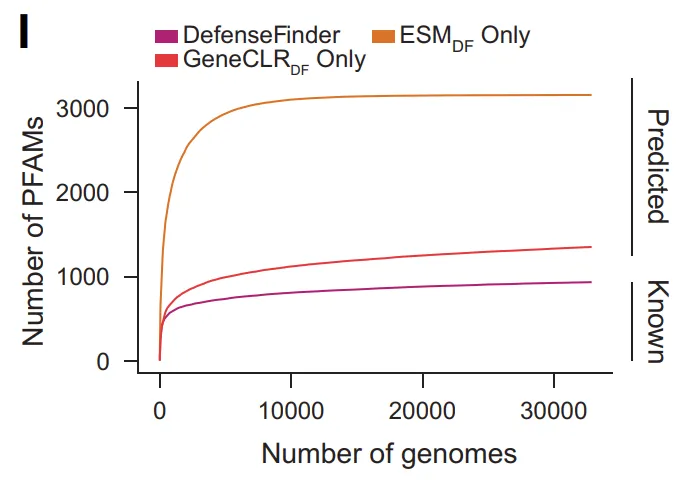
Au niveau de la diversité moléculaire, GeneCLR_DF a étendu le nombre de familles Pfam impliquées dans la défense de 934 à 3 154 (soit environ 151 TP3T parmi toutes les familles Pfam). Parallèlement, plus de 400 000 familles de protéines prédites ne possédaient aucune annotation Pfam, et moins de 51 TP3T étaient répertoriées dans DefenseFinder ; plus de 3 500 familles d’opérons étaient composées exclusivement de protéines sans domaine connu. Ces résultats indiquent que…Une grande partie de l'espace moléculaire de la défense antiphage n'a pas encore été caractérisée de manière systématique.
L'apprentissage profond permet un bond en avant dans l'efficacité de la découverte des défenses antiphages
Les cadres de prédiction des systèmes antiphages basés sur l'apprentissage profond et les atlas immunitaires antiviraux bactériens qui en découlent ouvrent la voie à une recherche plus évolutive dans ce domaine : on passe de découvertes ponctuelles, fondées sur des cas isolés, à une exploration systématique basée sur la reconnaissance de formes. Ce changement améliore non seulement l'efficacité de la découverte de nouveaux mécanismes de défense, mais rapproche également la recherche académique et les applications industrielles.
Dans le milieu universitaire, cette approche s'est rapidement développée. De nombreux instituts de recherche ont commencé à combiner l'apprentissage automatique et l'analyse génomique pour tenter d'identifier les systèmes de résistance aux phages à plus grande échelle. Par exemple,Le modèle DefensePredictor, développé par une équipe du MIT,En s'appuyant sur la logique de modélisation des modèles de langage protéique et en intégrant les informations de séquence génique et de contexte génomique, une identification très sensible des protéines antiphages a été obtenue. Le modèle a été entraîné sur environ 17 000 génomes de référence procaryotes et a identifié environ 821 nouveaux systèmes de défense TP3T lors de tests indépendants, validant ainsi la faisabilité de la « découverte de fonctions inconnues par reconnaissance de formes ».
Titre de l'article : DefensePredictor : un modèle d'apprentissage automatique pour la découverte des systèmes immunitaires procaryotes
Lien vers l'article :
https://www.science.org/doi/10.1126/science.adv7924
Dans l'industrie, les technologies connexes sont également mises en œuvre rapidement. Face à l'aggravation de la résistance aux antibiotiques, les bactériophages et leurs technologies dérivées regagnent en importance et constituent une voie essentielle pour remplacer ou compléter les antibiotiques traditionnels. Locus Biosciences, société biopharmaceutique en phase clinique, a développé une plateforme basée sur des bactériophages modifiés, combinant apprentissage automatique et biologie synthétique pour concevoir LBP-EC01, un traitement candidat contre Escherichia coli multirésistant, améliorant ainsi la précision et la contrôlabilité de la phagothérapie.
Parallèlement, Micreos privilégie une approche plus applicative, axée sur l'industrialisation des bactériophages et des endosomalines. Son produit Listex est utilisé dans l'industrie agroalimentaire pour inhiber la contamination par Listeria et a reçu des autorisations réglementaires dans plusieurs pays ; Staph Efekt exploite les propriétés bactéricides spécifiques des endosomalines dans les soins de la peau. Cette approche met l'accent sur la mise en œuvre fonctionnelle, transformant les mécanismes antiphages en produits concrets et utilisables, plutôt que de se limiter à la recherche en laboratoire.
Globalement, des modèles algorithmiques à la vérification expérimentale, puis aux applications industrielles, la recherche sur les antiphages forme progressivement une chaîne plus complète. Il est prévisible qu'avec l'accumulation de données et l'itération des modèles, cette approche, partant du calcul, se poursuivant par la vérification expérimentale et guidée par les applications, continuera de favoriser une compréhension plus approfondie des systèmes immunitaires bactériens et de traduire plus efficacement ces découvertes en solutions concrètes.
Liens de référence :
https://mp.weixin.qq.com/s/usrVEOeBD5gphhslZahLCA
https://mp.weixin.qq.com/s/Pxlh69TXSr8ffAp_ul3URw








