Command Palette
Search for a command to run...
ICLR 2026 | NVIDIA/Université d'Oxford Et d'autres Proposent Une Méthode De Génération De Liants Protéiques Au Niveau Atomique Avec Des Performances De Pointe (SOTA).

Dans le domaine de la biologie computationnelle,Concevoir des protéines capables de se lier précisément à des cibles spécifiques est l'un des problèmes les plus critiques et les plus difficiles.Elle est non seulement directement liée à des domaines clés tels que le développement de médicaments, la biothérapie et l'ingénierie enzymatique, mais elle détermine également la limite supérieure de l'efficacité humaine dans l'intervention sur les maladies complexes et la bioproduction.
D'un point de vue moléculaire, la liaison d'une protéine à une cible est essentiellement un problème structurel tridimensionnel :La composition en acides aminés, la conformation spatiale et les interactions intermoléculaires de l'interface déterminent collectivement l'affinité et la spécificité de la liaison.Par conséquent, presque toutes les méthodes de conception de liants reviennent finalement à la variable centrale de la « structure », en utilisant l'analyse ou la prédiction de la structure pour guider la construction moléculaire.
Ces dernières années, l'introduction de l'apprentissage automatique a profondément modifié ce paradigme. Grâce aux avancées réalisées dans les modèles de prédiction et de génération de structures, la recherche s'affranchit progressivement de sa forte dépendance aux structures expérimentales, passant de l'« analyse des structures » à la « génération des structures ». Il devient ainsi possible de concevoir des liants de novo et de réduire considérablement les coûts et les délais de R&D.
Cependant, en termes de méthodologie, la conception actuelle de liants pilotée par l'IA présente encore une divergence claire :L'un de ces types est constitué des méthodes génératives, représentées par RFDiffusion.Il s'appuie sur un entraînement à grande échelle pour générer directement des structures candidates, mais manque de la capacité de s'ajuster de manière flexible pendant la phase d'inférence ;Un autre type est la méthode illusoire, représentée par BindCraft.L'optimisation par gradient via l'évaluation de prédicteurs de structure offre une grande flexibilité, mais manque de connaissances a priori génératives, ce qui rend difficile l'exploration d'espaces structurels entièrement nouveaux. Cette « séparation entre génération et optimisation » contraste avec le paradigme unifié « modèles pré-entraînés + extensions de calcul au moment de l'inférence » déjà établi dans les domaines du traitement automatique du langage naturel et du traitement d'images.
C'est dans ce contexte queUne équipe de recherche conjointe de NVIDIA, de l'Université d'Oxford, de l'Institut québécois d'intelligence artificielle et d'autres institutions a proposé le cadre Proteina-Complexa (ci-après dénommé Complexa).Visant à combler le fossé entre les méthodes génératives et illusionnistes, cette approche unifie le modèle génératif de base et les mécanismes d'optimisation lors de l'inférence au sein d'un seul système. Elle repose sur le pré-entraînement de Teddymer.Complexa permet la conception de novo d'agents de liaison de pointe sans nécessiter d'étapes supplémentaires de reconception de séquence.En adaptant la technique de mise à l'échelle du temps de test du modèle de diffusion à ce cadre, la génération et l'optimisation sont directement unifiées, surpassant ainsi les méthodes traditionnelles basées sur l'illusion en termes de performances.
Les résultats de recherche associés, intitulés « Mise à l'échelle de la conception de liants protéiques atomistiques avec pré-entraînement génératif et calcul au moment du test », ont été acceptés pour ICLR 2026.
Points saillants de la recherche :
* Cette étude propose Complexa, qui étend La-Proteina à la conception de liants, utilise Teddymer et réalise une optimisation efficace du temps d'inférence accélérée par des a priori génératifs.
* Obtention de taux de réussite de pointe en simulation informatique pour les cibles protéiques et de petites molécules ainsi que pour la conception d'enzymes, sans nécessiter de reconception de séquence.
Adresse du document :
https://openreview.net/forum?id=qmCpJtFZra
Suivez notre compte WeChat officiel et répondez « Complexa » en arrière-plan pour obtenir le PDF complet.
Jeux de données : De « l’enrichissement d’unités uniques » à la « reconstruction complexe »
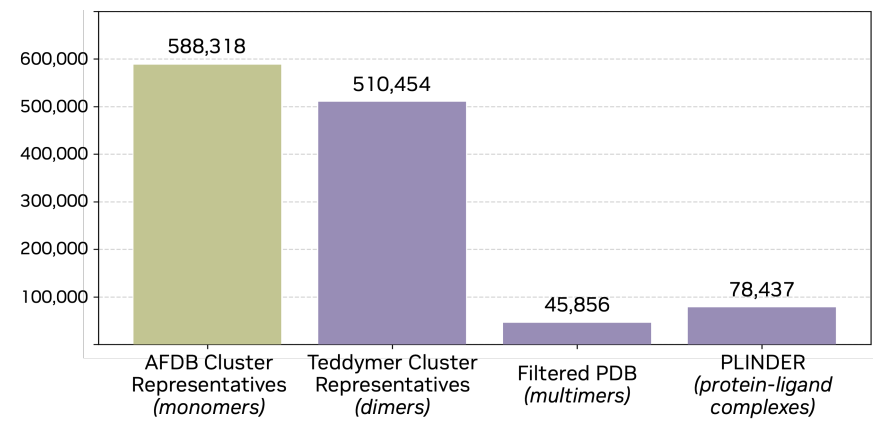
Une limitation fondamentale des modèles de génération de ligands réside dans les données. Idéalement, le modèle nécessite une grande quantité de données de complexes « agent de liaison-cible » pour son apprentissage. Or, en réalité, ce type de données provient principalement de bases de données de protéines analysées expérimentalement (PDB), dont la taille est limitée et les échantillons de haute qualité encore plus rares. Quant à la base de données AlphaFold (AFDB), plus vaste, elle fournit une quantité massive de structures protéiques, mais la quasi-totalité d'entre elles sont des structures monomériques, dépourvues d'informations sur les complexes.Ce décalage structurel entre « l'abondance des composants individuels et la rareté des composants complexes » limite directement la capacité du modèle à être entraîné à grande échelle.
La principale avancée de cette étude réside dans une compréhension renouvelée de la structure interne d'AFDB. La plupart des protéines d'AFDB sont des protéines multidomaines, et l'outil de segmentation de domaines TED leur fournit une annotation précise. Une analyse plus poussée a révélé que les interactions entre différents domaines au sein d'une même protéine présentent des caractéristiques statistiquement similaires à celles observées dans les complexes multi-brins. Cette observation conduit à un changement important :Les structures monomères ne sont pas des « données inutiles » en soi, mais peuvent être réinterprétées comme une source potentielle de données pour les structures complexes.
Partant de cette observation, l'étude propose une méthode de « construction de multimères artificiels ».En décomposant les protéines multidomaines en domaines indépendants et en les traitant comme des chaînes différentes, une structure de type complexe peut être construite au sein du monomère.Le processus débute avec AFDB50, en sélectionnant les protéines annotées TED, puis en les divisant en « pseudo-multimères ». Les dimères sont ensuite extraits et filtrés selon leur proximité spatiale, seuls les échantillons entièrement annotés étant conservés. Après un regroupement par clustering visant à éliminer les redondances, environ 3,5 millions de clusters de dimères sont obtenus. Cet ensemble de données, nommé Teddymer, ne se contente pas d'accroître la taille des données, mais transforme l'avantage des monomères en une offre de complexes grâce à une réorganisation structurale.
Au cours du processus d'entraînement, comme le montre la figure ci-dessous, l'étude ne s'est pas appuyée sur une seule source de données, mais a intégré les données de monomères AFDB, les données de construction Teddymer, les données de complexes expérimentaux PDB et les données protéine-ligand PLINDER, permettant au modèle d'établir une représentation unifiée entre la structure des monomères, la structure des complexes et les interactions des petites molécules, prenant ainsi en compte à la fois les capacités génératives et de généralisation.
Complexa : Un cadre atomique complet pour la génération d'agents de liaison aux protéines
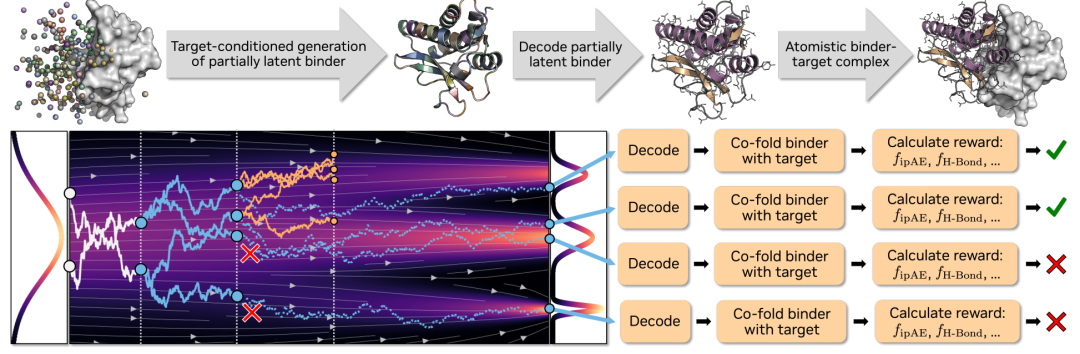
En termes de conception de modèles, le changement fondamental apporté par Complexa ne réside pas simplement dans une « capacité de génération accrue », mais plutôt dans un changement de cible générative, passant des « structures protéiques complètes » aux « agents de liaison à des interfaces spécifiques ». Basé sur La-Proteína, son avantage réside dans sa capacité à générer à l'échelle atomique entière.Parallèlement, grâce à l'architecture efficace du Transformer, il évite les modules à coût de calcul élevé présents dans les modèles structurels traditionnels, ce qui lui confère une bonne évolutivité dans les scénarios d'échantillonnage à grande échelle.
S’appuyant sur ce constat, l’étude introduit un mécanisme de génération conditionné par les points cibles et les zones d’interaction. Ce mécanisme empêche le modèle de générer des complexes complets, se limitant à la partie liant, et repose explicitement sur les informations cibles lors du processus de génération. Plus précisément,Le modèle de correspondance de flux est responsable de la génération de structures sous contraintes conditionnelles, tandis que l'auto-encodeur n'est utilisé que pour l'encodage et le décodage des liants monomères, réduisant ainsi la complexité de la modélisation tout en maintenant la puissance expressive.
Pour permettre au modèle de comprendre efficacement les informations cibles, une conception systématique de la représentation des entrées a été mise en œuvre. Les cibles protéiques ont été encodées à l'aide de la méthode Atom37, les coordonnées 3D au niveau des résidus, les types d'acides aminés et les informations sur les points chauds d'interface étant uniformément intégrés au modèle. Ces points chauds indiquent les régions de liaison potentielles. Lors de l'entraînement, ces points chauds ont été extraits d'interfaces réelles, tandis que lors de l'inférence, ils ont été utilisés comme a priori ou obtenus par prétraitement. Pour les cibles de petites molécules, le modèle a encodé le type, la charge et les coordonnées spatiales au niveau atomique, et ces informations ont été intégrées, avec la représentation de l'agent de liaison, au Transformer pour la modélisation conjointe.
En ce qui concerne les objectifs de formation,Une amélioration clé réside dans l'introduction d'un bruit de translation global aléatoire dans les coordonnées du liant, forçant ainsi le modèle à apprendre la capacité de localisation spatiale des molécules.Ceci n'est pas crucial pour la génération de monomères, mais constitue une capacité essentielle qui détermine la qualité de la génération pour les tâches exigeant un positionnement précis du liant à l'interface cible. L'ensemble du processus d'apprentissage adopte une stratégie progressive, passant de la modélisation des monomères à la génération de la structure générale, puis à l'apprentissage spécifique au liant. Parallèlement, le surapprentissage est contrôlé par LoRA, et l'auto-encodeur au niveau des monomères est réutilisé tout au long du processus afin de préserver la simplicité de l'architecture.
Au stade de l’inférence,Complexa introduit en outre un mécanisme d'« expansion des calculs en temps de test », qui combine le processus de génération avec l'optimisation de la recherche.En augmentant le nombre d'échantillons, en introduisant la recherche par faisceaux ou la recherche arborescente Monte Carlo, le modèle peut améliorer continuellement la qualité de sa génération tout en bénéficiant d'un budget de calcul plus important. Cette conception permet aux capacités du modèle de s'étendre dynamiquement pendant l'inférence, au lieu d'être entièrement limitées à la phase d'entraînement.
Taux de réussite plus élevé, vitesse accrue et évolutivité renforcée
Pour valider les capacités du modèle, cette étude a conçu une série d'expériences, des plus simples aux plus complexes. La question centrale est la suivante : Complexa surpasse-t-il non seulement les autres modèles en termes de performances de base, mais continue-t-il également à améliorer ses performances à mesure que les ressources de calcul augmentent ?
En termes de capacités de production de base,Qu’il s’agisse de cibler des protéines ou de petites molécules, Complexa surpasse nettement les méthodes existantes, affichant des taux de réussite plus élevés et des vitesses d’échantillonnage plus rapides.Parallèlement, la nouveauté des structures générées est nettement améliorée. Plus important encore, le modèle peut produire directement des séquences de haute qualité sans recourir à des outils tels que ProteinMPNN pour la conception secondaire, simplifiant ainsi l'ensemble du processus.
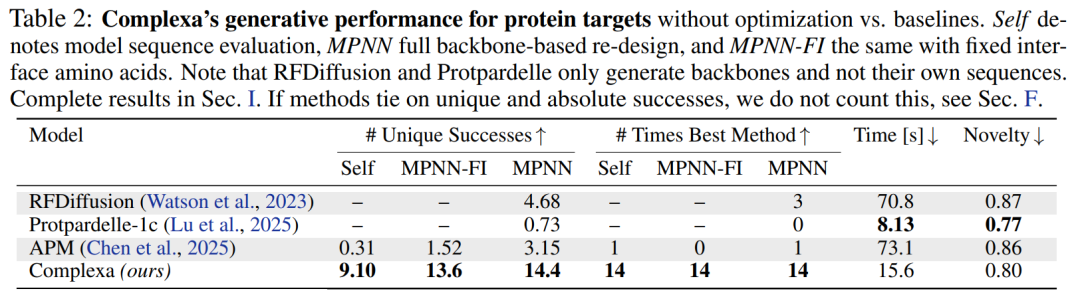
En termes de contrôlabilité structurelle,L'étude introduit des étiquettes conditionnelles, permettant au modèle de contrôler explicitement le type de structure générée.Par exemple, le choix entre les hélices α et les replis β peut atténuer efficacement le problème de la structure unique des modèles génératifs précédents et améliorer considérablement la diversité structurale.
Dans les expériences d'extension de calcul pendant la phase d'inférence, les résultats montrent que sur des tâches simples, le simple fait d'augmenter le nombre d'échantillons suffit à surpasser toutes les méthodes de référence, tandis que sur des tâches complexes, l'introduction de stratégies de recherche plus avancées (telles que la recherche par faisceau et la recherche arborescente Monte Carlo) amplifie encore l'avantage.Cela indique que les performances du modèle peuvent continuer à s'améliorer à mesure que le budget de calcul augmente.Comme le montre la figure ci-dessous
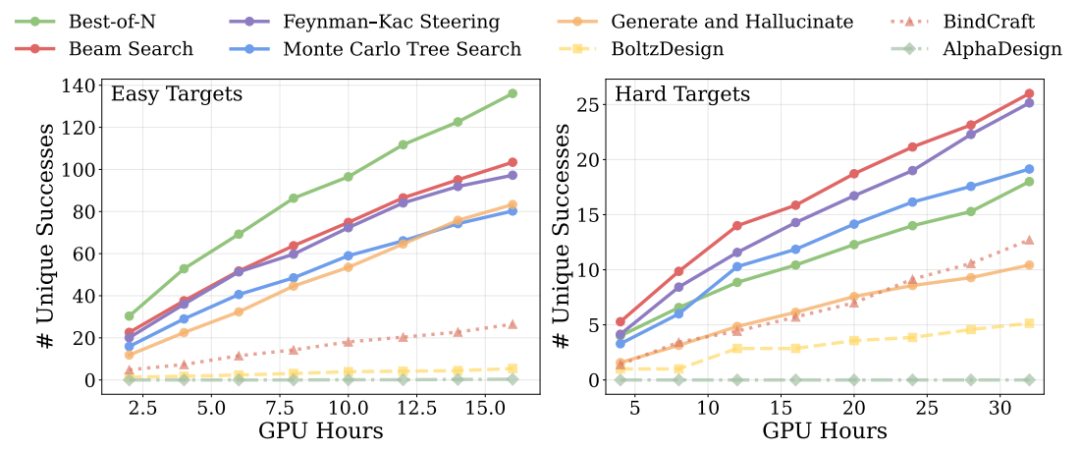
En ce qui concerne la plausibilité physique, des recherches supplémentaires sont nécessaires pour optimiser les liaisons hydrogène interfaciales et les indices énergétiques associés.Le modèle s'est avéré capable non seulement de générer des liants structurellement solides, mais aussi de les optimiser au niveau d'interaction le plus fin.Cela améliore la stabilité de la liaison.
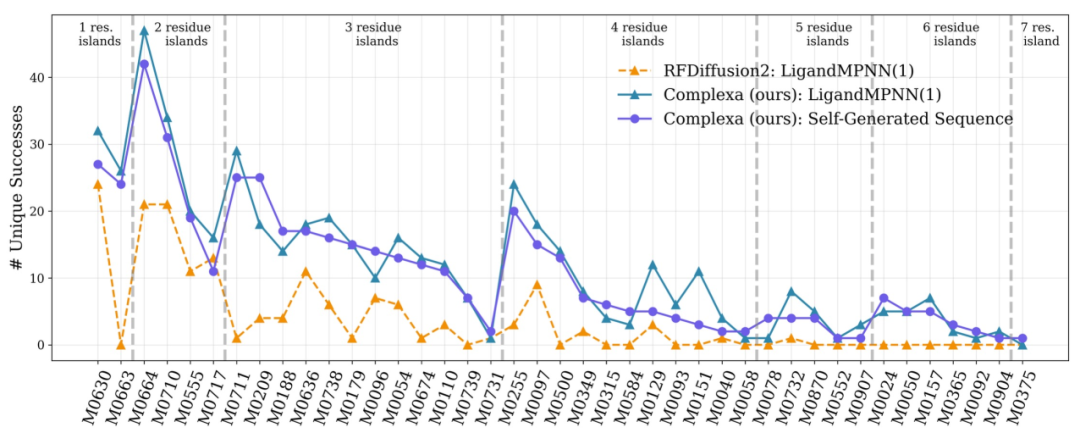
Dans les tâches cibles multi-chaînes plus complexes, les méthodes existantes ne peuvent pas obtenir de solutions efficaces avec des budgets de calcul limités.Complexa a généré avec succès des candidats de haute qualité après avoir augmenté ses ressources de calcul, démontrant ainsi son évolutivité face à des problèmes complexes.Enfin, les tests effectués sur différentes tâches, telles que la conception d'enzymes, sont présentés dans la figure ci-dessous. Le cadre proposé possède une bonne capacité de généralisation et peut être étendu de la conception de liants à un plus large éventail de problèmes d'ingénierie des protéines.
Changement de paradigme dans la conception des protéines par l'IA
Ces dernières années, la conception d'agents de liaison aux protéines pilotée par l'IA a rapidement progressé de la théorie à la pratique, et le lauréat du prix Nobel David Baker et son équipe demeurent des figures de proue dans ce domaine. En 2025, leur équipe a publié plusieurs études dans la revue Science.Le système a vérifié la faisabilité de la conception de liants pMHC hautement spécifiques basés sur la diffusion RF.Les travaux connexes ont ciblé 11 catégories de maladies, ont généré avec succès des protéines de liaison capables d'amener les lymphocytes T à reconnaître les tumeurs et ont vérifié la précision de la conception à l'échelle atomique à l'aide de la cryo-microscopie électronique, marquant ainsi le début de la vérifiabilité pour la conception de l'IA.
Parallèlement, l'équipe du MIT a exploré une approche plus intégrée dans le modèle BoltzGen, unifiant la prédiction de structure et la génération d'agrégats dans un seul modèle atomique et utilisant des représentations géométriques continues au lieu de la modélisation discrète traditionnelle.Dans des expériences ciblant 26 cibles, le 66% a atteint une affinité nanomolaire en tant que liant.Elle maintient également un taux de réussite élevé sur les cibles hors distribution, démontrant une certaine capacité de généralisation.
L'industrie se concentre davantage sur la mise en œuvre technique de ces capacités. Début 2026, Bayer et Cradle ont entamé une collaboration de trois ans afin d'intégrer leur plateforme d'ingénierie des protéines basée sur l'IA au processus de développement des anticorps. Cette plateforme a été appliquée dans plus de 50 projets, raccourcissant considérablement les cycles de développement et favorisant un processus itératif en boucle fermée de « conception-test-apprentissage ». Cela témoigne de la transition de l'IA d'un outil auxiliaire à une capacité fondamentale au sein du processus de R&D.
De manière générale, la compétition en conception de protéines évolue : on passe de la performance d’un modèle unique à l’efficacité et à l’évolutivité au niveau du système. Le monde académique continue de repousser les limites des capacités de modélisation, tandis que l’industrie les oriente vers un processus de R&D stable et réutilisable. La conception de protéines par IA entre ainsi dans une phase plus pratique : l’enjeu n’est plus de savoir « si elle est possible à concevoir », mais « si elle peut l’être de manière continue et efficace ».
Liens de référence :
1.https://news.bioon.com/article/00bf92186439.html
2.https://mp.weixin.qq.com/s/1zKXUQtXgCJ7GA1_OUEShg
3.https://www.bayer.com/en/us/news-stories/ai-enabled-antibody-discovery-and-optimization