Command Palette
Search for a command to run...
Le MIT a Proposé VibeGen, Le Premier Modèle De Génération De Protéines Dynamique De Bout En Bout, Qui Réalise Une Correspondance Bidirectionnelle Entre La Séquence Et La vibration.
Les protéines sont les molécules fonctionnelles essentielles des systèmes vivants, mais leurs fonctions ne sont pas uniquement déterminées par leur structure statique ; elles résultent plutôt de leur dynamique conformationnelle en constante évolution. Au sein d’un paysage énergétique complexe, les protéines maintiennent un équilibre dynamique dans des conditions physiologiques grâce à des mouvements multi-échelles allant de la femtoseconde à la milliseconde, ce qui en fait de véritables machines moléculaires.
C’est pourquoi une dynamique protéique anormale est étroitement associée à diverses maladies. Par exemple, la protéine suppresseur de tumeur p53 fonctionne selon un mécanisme dépendant de sa plasticité conformationnelle, et les mutations oncogènes affaiblissent cette capacité ; les mutations du gène CFTR, quant à elles, induisent la mucoviscidose en perturbant la dynamique d’ouverture et de fermeture des canaux ioniques. Ces faits indiquent que…Le « mouvement » des protéines est en soi un déterminant important de leur fonction.Par conséquent, la compréhension et la conception des protéines dans une perspective dynamique deviennent une direction de pointe en biologie structurale et en bioingénierie.
Au cours des dernières décennies, les chercheurs ont mis au point des techniques expérimentales telles que la résonance magnétique nucléaire (RMN), la spectrométrie de masse par échange hydrogène-deutérium (HDEMS) et la cryo-microscopie électronique (cryo-ME), ainsi que des méthodes de calcul comme les simulations de dynamique moléculaire et l'analyse des modes normaux de vibration (VMS), afin de caractériser la dynamique des protéines. Cependant, ces méthodes sont soit trop complexes pour être appliquées à grande échelle, soit trop coûteuses en ressources de calcul et trop limitées en temps, ce qui les rend inadaptées aux études à grande échelle.
Ces dernières années, l'apprentissage profond et l'IA générative ont ouvert de nouvelles perspectives à la recherche sur les protéines. Des modèles comme AlphaFold2 ont permis d'obtenir des prédictions de structure de haute précision, et certaines méthodes peuvent également prédire les structures secondaires, les sites de liaison et même les caractéristiques vibrationnelles. Cependant,La plupart des méthodes existantes restent encore au niveau de la « structure ou d'une propriété unique », sans modélisation systématique de la dynamique intrinsèque.Dans le domaine de la conception, des frameworks tels que RFdiffusion et AlphaFold3 traitent encore les structures comme des corps rigides approximatifs et n'ont pas encore véritablement introduit de contraintes dynamiques. Par conséquent, établir une correspondance unifiée entre « séquence-structure-dynamique-fonction » et parvenir à une conception contrôlable basée sur la dynamique demeure un défi majeur.
Récemment,Une équipe de recherche conjointe du MIT et de l'Université Carnegie Mellon a proposé VibeGen, un agent intelligent générateur de protéines.En combinant la génération de séquences et la prédiction de la dynamique vibrationnelle, la conception de protéines de novo a été réalisée. Les résultats montrent que les protéines conçues par cet agent génératif peuvent non seulement se replier en structures stables et inédites, mais aussi reproduire les caractéristiques de distribution des amplitudes vibrationnelles cibles au niveau de la chaîne principale.
Les résultats de cette recherche, intitulée « VibeGen : Conception de protéines de novo de bout en bout et autonome pour une dynamique sur mesure utilisant un modèle de diffusion du langage », ont été publiés dans Matter.
Adresse du document :
https://www.cell.com/matter/abstract/S2590-2385(26)00069-X
Base de données de dynamique des protéines basée sur les modes de vibration normaux de basse fréquence
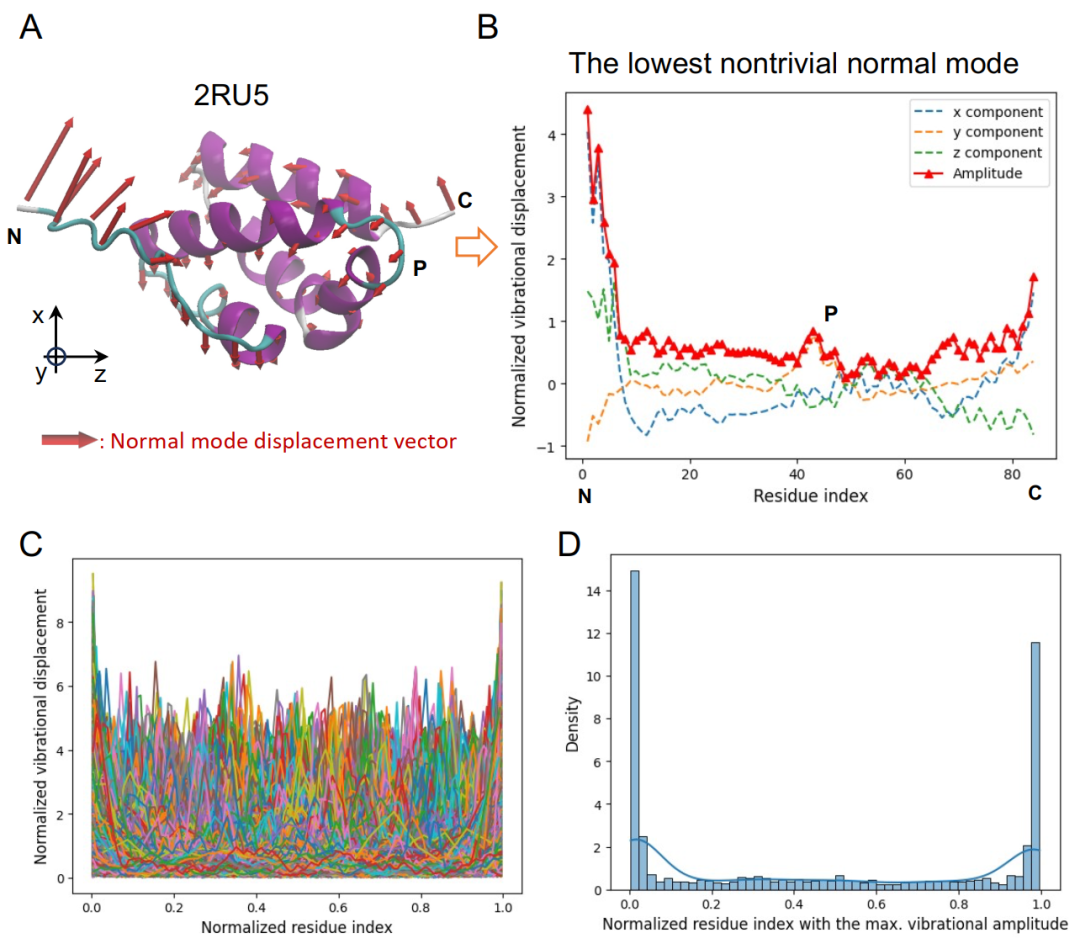
Pour constituer la base de données,Les chercheurs ont passé au crible des protéines à chaîne unique d'une longueur n'excédant pas 126 acides aminés à partir de la base de données de protéines (PDB) mise à jour en janvier 2024.La structure a été nettoyée et complétée à l'aide d'outils tels que VMD, MMTSB et SCWRL4. Une minimisation d'énergie a ensuite été réalisée à partir du champ de force CHARMM, et les informations modales ont été calculées par la méthode des modes de vibration normaux par blocs. Après suppression des six premiers modes de corps rigide représentant la translation et la rotation globales, le mode non trivial de plus basse fréquence a été sélectionné pour l'analyse ultérieure.
S’appuyant sur ces résultats, l’étude a extrait les modes de déplacement des atomes Cα de chaque résidu de la chaîne principale, construisant ainsi un vecteur de forme de mode vibrationnel normal. Les résultats ont révélé une distribution hétérogène des déplacements vibrationnels : des amplitudes plus importantes aux extrémités de la chaîne et dans les régions peu structurées, tandis que les vibrations étaient restreintes dans les régions denses telles que les hélices α et les feuillets β. Les régions en coude et enroulées présentaient des pics locaux dus à leur plus grande flexibilité. Afin d’éliminer l’influence des différences de longueur, les vecteurs ont été normalisés, ce qui en fait des descripteurs dynamiques indépendants des coordonnées.
final,Les chercheurs ont constitué un ensemble de données contenant 12 924 brins simples de protéines.L'analyse révèle une grande diversité des modes de vibration basse fréquence, avec des amplitudes maximales concentrées aux extrémités de la chaîne. L'ensemble de données a été divisé en ensembles d'entraînement et de test selon un ratio de 9:1 pour l'entraînement et l'évaluation ultérieurs du modèle génératif.
VibeGen : Conception de protéines de novo de bout en bout basée sur un modèle de diffusion du langage
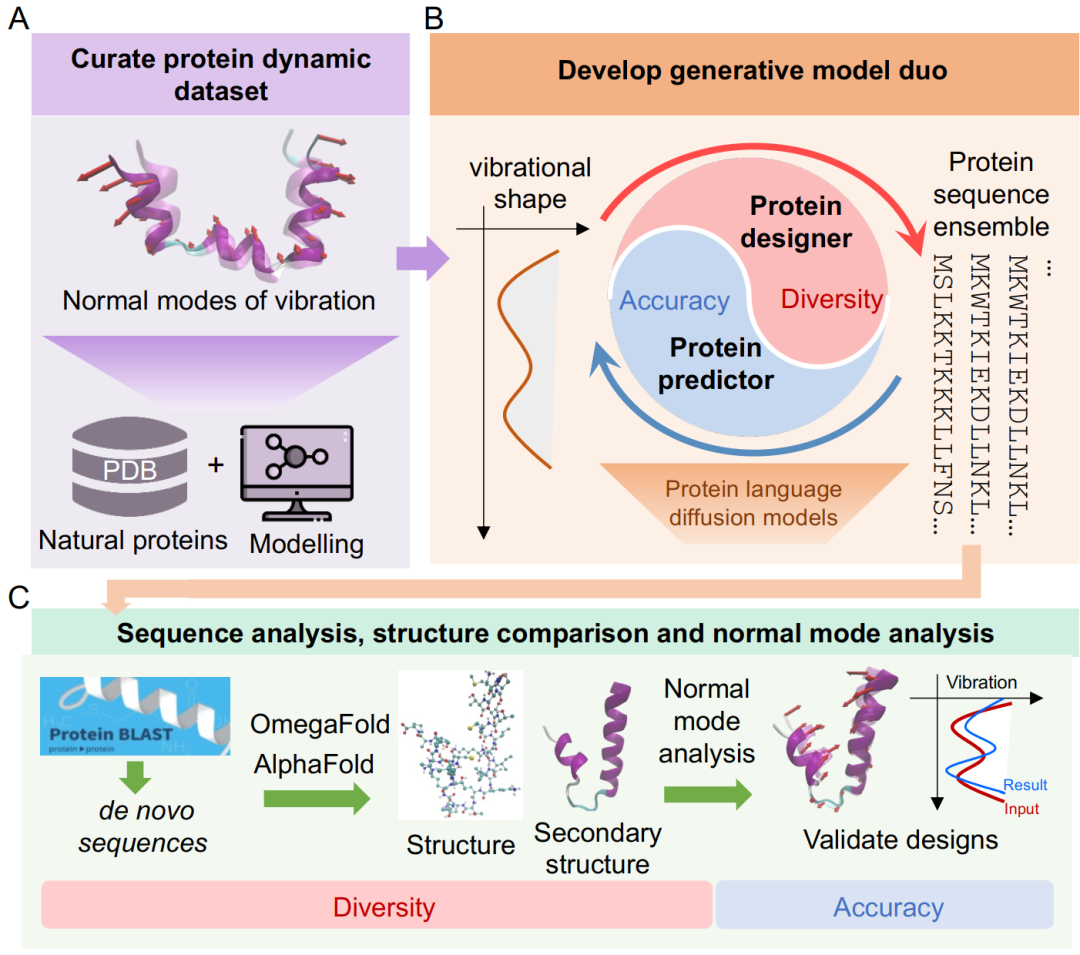
La principale difficulté de cette étude réside dans le fait que la forme des modes de vibration normaux est déterminée par la structure tridimensionnelle complexe et les propriétés élastiques des protéines, et qu'il n'existe pas de relation directe entre la séquence et la dynamique. Par ailleurs, l'information relative à un seul mode est fortement dégénérée, et différentes séquences peuvent correspondre à des caractéristiques dynamiques similaires, ce qui rend le problème de conception inverse particulièrement complexe.
Pour relever ces défis, cette étude a d'abord extrait les principales caractéristiques dynamiques d'un grand nombre de protéines à partir d'une base de données de protéines (PDB) grâce à une analyse des modes de vibration normaux et à des simulations de dynamique moléculaire tout atome. Sur cette base,Les chercheurs ont construit deux modèles de diffusion de langage protéique collaboratifs : un module de conception de protéines (PD) et un module de prédiction (PP).Ils sont respectivement responsables de la prédiction directe et de la conception inverse entre la séquence et l'espace des modes vibrationnels normaux. Les deux modules ont des structures similaires, reposant toutes deux sur la combinaison d'un modèle de langage protéique pré-entraîné (pLM) et d'un modèle de diffusion.
La tâche du module de conception est de générer une séquence basée sur les caractéristiques dynamiques cibles.Au cours du processus de débruitage, le modèle de diffusion intègre des informations sur les conditions dynamiques à travers de multiples canaux et génère progressivement des séquences conformes aux caractéristiques cibles dans l'espace latent.Le module de prédiction possède une structure symétrique et déduit la forme du mode de vibration normal à partir de la séquence d'entrée. Il utilise également plusieurs représentations de séquences issues du modèle de langage pré-entraîné afin d'optimiser les résultats de prédiction.
Les deux modules sont entraînés indépendamment, et ensemble ils forment un système collaboratif en boucle fermée de « génération-évaluation-sélection » pendant la phase de déploiement.Le module de conception génère d'abord des séquences candidates, et le module de prédiction évalue leurs performances dynamiques en temps réel.Les chercheurs peuvent filtrer les résultats en fonction de leurs besoins en matière de précision ou de diversité, et répéter les itérations autant de fois que nécessaire jusqu'à l'obtention d'une séquence satisfaisante.
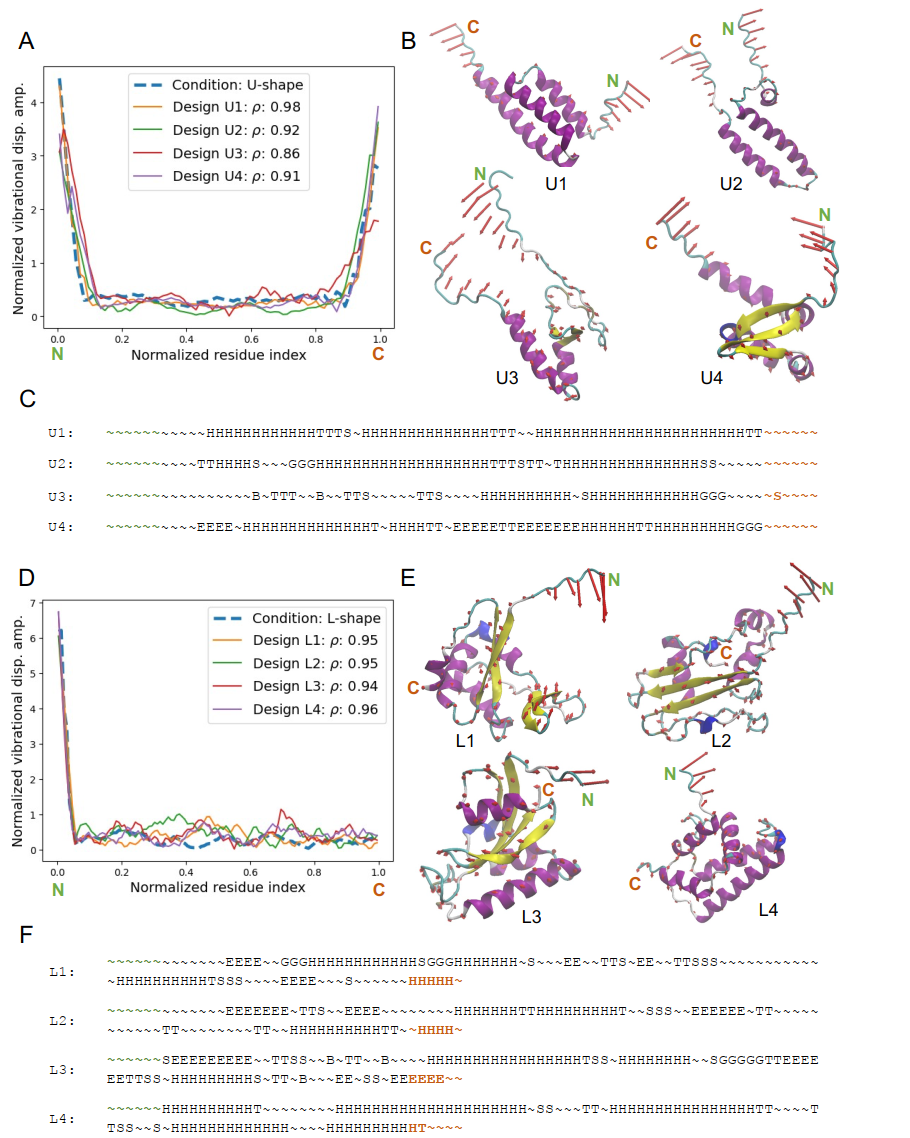
Les performances du modèle ont été validées sur l'ensemble de test. Pour différentes formes de modes de vibration normaux typiques, notamment en L, en U et en W, la protéine générée par le modèle a été validée par une analyse réelle des modes de vibration normaux, et sa forme vibratoire correspondait parfaitement à la cible de conception. Des indicateurs quantitatifs tels que le coefficient de corrélation de Pearson et l'erreur relative L2 ont montré que…Cette méthode permet d'obtenir une conception de haute précision malgré des contraintes dynamiques complexes.
D'un point de vue structurel, la formation des protéines présente une correspondance cinétique claire : les régions avec des vibrations plus fortes ont tendance à être des pelotes aléatoires ou des fragments flexibles, tandis que les régions avec des vibrations restreintes ont tendance à former des structures stables telles que des hélices α ou des feuillets β.Cela démontre que le modèle a efficacement saisi la relation intrinsèque entre la structure et la dynamique.
Au niveau de l'implémentation du modèle, les modules de conception et de prédiction utilisent un modèle pré-entraîné de taille moyenne (150 millions de paramètres) issu de la série ESM-2 comme modèle de prédiction (pLM), afin d'optimiser le compromis entre efficacité de calcul et performance. Le modèle de diffusion intègre des informations conditionnelles au processus de débruitage via plusieurs canaux d'un réseau en U et est entraîné indépendamment à l'aide de l'optimiseur Adam.
Une avancée majeure en matière de précision et de nouveauté
Pour évaluer la performance du modèle, l'étude a mené des analyses expérimentales multidimensionnelles. L'analyse de la diversité a montré que…Pour un même objectif dynamique, le modèle peut générer plusieurs schémas de conception avec des structures différentes mais la même fonction.Prenons comme exemples les modes de vibration normaux en forme de U et de L : les protéines conçues présentent toutes une structure « noyau dense + extrémités ouvertes ». Les extrémités sont des structures en pelote statistique, correspondant aux régions de forte amplitude ; le noyau peut être constitué de diverses structures, telles que des faisceaux d’hélices α ou des structures hybrides à repliement hélicoïdal, correspondant aux régions de faible amplitude. Cette diversité provient principalement du degré de liberté dans le choix de la structure au sein des régions de faible vibration, et le modèle capture et exploite avec succès ces « solutions multiples ».
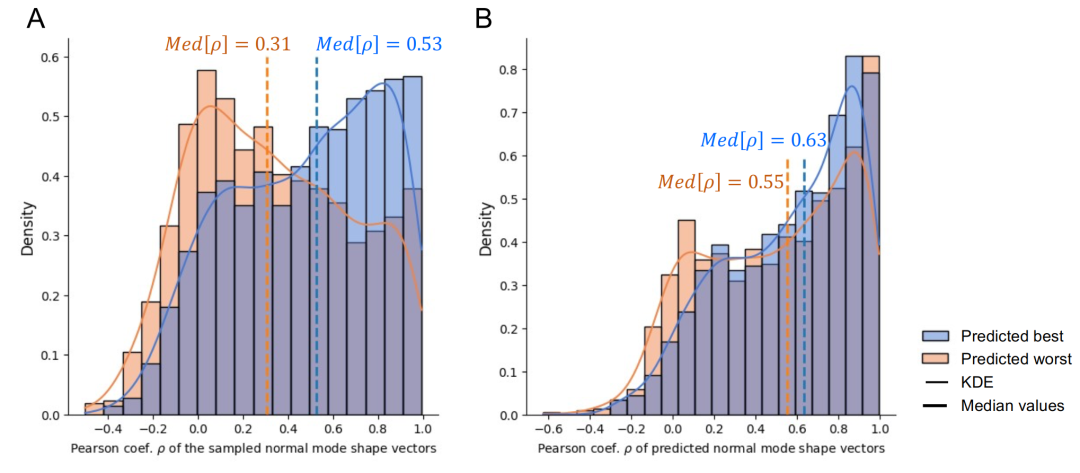
L'efficacité du module de prédiction a été vérifiée par des expériences comparatives. Comme le montre la figure ci-dessous, lors de la sélection des meilleurs et des pires groupes de prédiction parmi un même ensemble de séquences candidates, la précision de conception réelle du premier était significativement supérieure à celle du second (coefficient de corrélation de Pearson médian de 0,53 contre 0,31), tandis que le module de prédiction a maintenu une précision stable pour les deux groupes. Ceci indique que…L'introduction d'un module de prédiction lors du processus de conception permet de sélectionner efficacement les séquences de haute qualité et de réduire la dépendance à une vérification physique coûteuse.
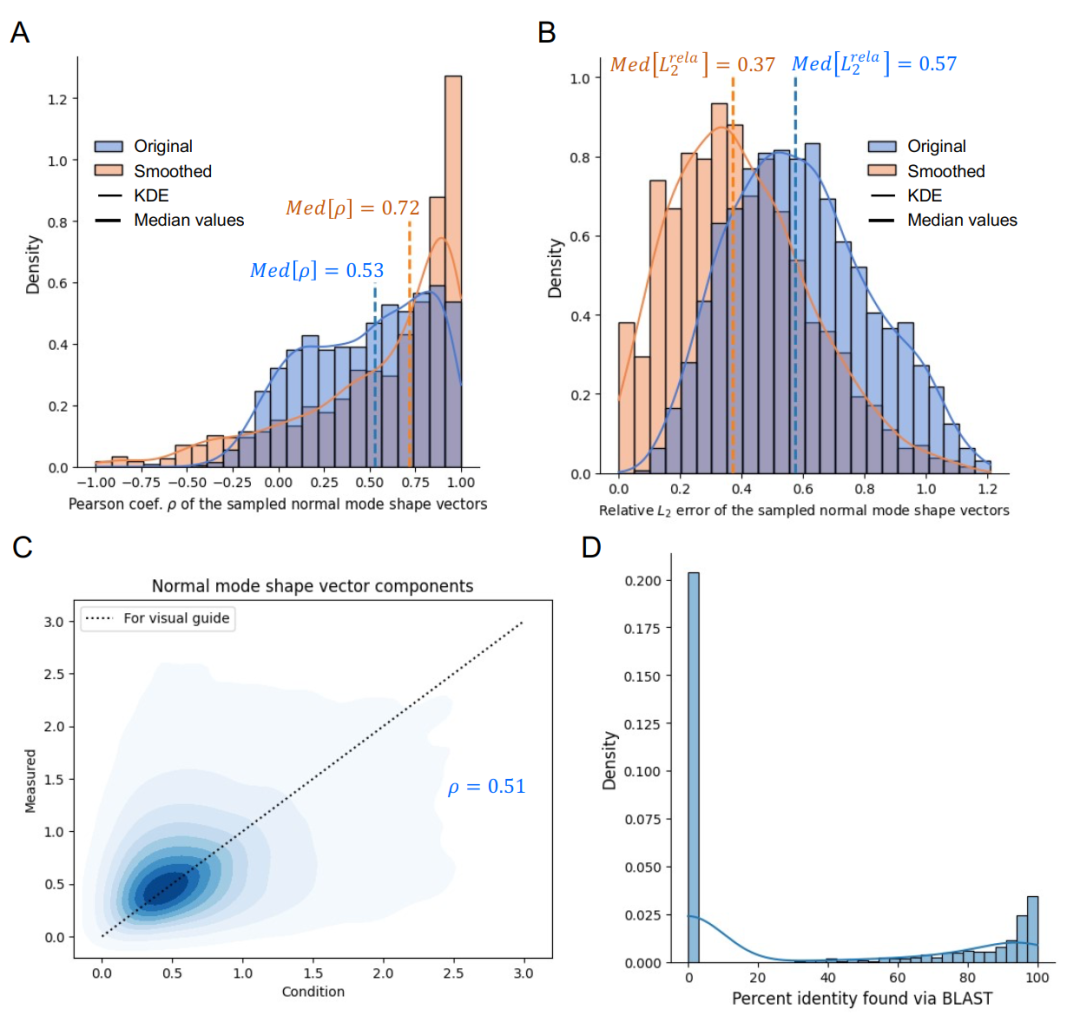
Les statistiques de performance globales sont basées sur 1 293 cas de test. Comme illustré dans la figure ci-dessous, le coefficient de corrélation médian entre le mode propre mesuré et la valeur cible est de 0,53, et l'erreur relative L2 médiane est de 0,57, ce qui reflète la difficulté inhérente à la conception de haute précision au niveau des résidus. Après filtrage passe-bas pour préserver la forme globale, le coefficient de corrélation médian passe à 0,72 et l'erreur médiane diminue à 0,37.Cela indique que le modèle est particulièrement performant pour reproduire le profil global des vibrations.Cette caractéristique revêt une importance biologique capitale pour la dynamique conformationnelle à grande échelle des protéines.
En termes de nouveauté, l'identité de séquence la plus élevée de BLAST présente une distribution bimodale, le pic principal correspondant aux séquences conçues de novo.Cela indique que le modèle est plus susceptible de générer de nouvelles séquences, élargissant ainsi la bibliothèque potentielle de solutions en matière de structure et de dynamique des protéines.
La corrélation entre la structure et la dynamique est systématiquement démontrée dans de multiples séries d'expériences : les structures denses telles que les hélices α et les replis β sont principalement distribuées dans les régions de faible amplitude, tandis que les régions de forte amplitude sont principalement des régions de boucle ou des boucles terminales.Le modèle a permis de reproduire avec succès cette loi physique et de contrôler la flexibilité locale grâce à des éléments structurels secondaires, démontrant ainsi une compréhension de la relation structure-dynamique.
Globalement, ce modèle atteint un bon équilibre entre précision, diversité et nouveauté dans la conception de protéines sous contraintes cinétiques, jetant ainsi les bases de conceptions fonctionnelles ultérieures plus complexes.
Combinaison de la génération de protéines d'agents intelligents et de la conception inverse de modes vibratoires normaux
La recherche sur la génération de protéines d'agents intelligents et la conception inverse basée sur les modes de vibration normaux devient un sujet de pointe dans le domaine de l'ingénierie des protéines, stimulant à la fois l'exploration académique et l'innovation industrielle.
Dans le milieu universitaire, de nombreuses équipes de recherche travaillent sans relâche sur ce sujet et ont obtenu des résultats novateurs. Certaines équipes ont optimisé le cadre collaboratif des agents intelligents.La combinaison de l'analyse des modes de vibration normaux avec un modèle de diffusion du langage protéique plus avancé atténue efficacement le problème de dégénérescence dans la conception inverse.Ce travail a permis de vérifier plus en détail la relation intrinsèque entre la forme des modes de vibration normaux et la structure secondaire et les propriétés dynamiques des protéines, fournissant ainsi un support théorique plus solide et une voie technique pour la conception de novo de protéines aux fonctions spécifiques.
Une autre équipe s'est concentrée sur l'allègement et la généralisation, en optimisant la taille des paramètres et la stratégie d'entraînement des modèles de langage protéique pré-entraînés, et en développant des modèles plus petits et plus faciles à généraliser.De plus, l'application de la conception inverse des modes de vibration normaux a été étendue à des domaines spécifiques tels que la conception des sites catalytiques enzymatiques et l'optimisation des agents de liaison aux protéines.Cela a jeté des bases solides pour la transformation industrielle ultérieure.
Par ailleurs, Google DeepMind a lancé AlphaProteo,Premier outil d'intelligence artificielle destiné à la conception de nouveaux adhésifs protéiques à haute résistance, il peut générer de nouveaux conjugués protéiques pour une variété de protéines cibles.En incluant le facteur de croissance endothélial vasculaire A (VEGF-A), associé aux complications du cancer et du diabète, le test a obtenu un taux de réussite expérimental plus élevé. Son affinité de liaison est 3 à 300 fois supérieure à celle des meilleures méthodes existantes, ce qui devrait accélérer le développement de médicaments anticancéreux et antiviraux, et ouvre également de nouvelles perspectives pour le développement de biocapteurs et l'amélioration de la résistance des cultures aux insectes.
D'autres entreprises se concentrent sur les points faibles du développement de médicaments et utilisent la technologie de conception inverse de la forme du mode vibratoire normal pour concevoir des médicaments protéiques ciblant des maladies spécifiques, raccourcissant ainsi le cycle de développement, réduisant les coûts et promouvant le développement de médicaments protéiques de manière plus précise et efficace.
Actuellement, l'optimisation continue, par la communauté académique, de la précision de conception et de la capacité de généralisation des modèles, conjuguée aux efforts constants de l'industrie pour accroître l'efficacité de mise en œuvre et les cas d'application, contribue à faire progresser la technologie de conception des protéines vers une précision, une efficacité et une diversité accrues. À l'avenir, à mesure que cette technologie gagnera en maturité, les méthodes de conception des protéines basées sur des agents intelligents et l'analyse des modes de vibration normaux devraient trouver des applications plus larges dans des domaines tels que la pharmacie, la production industrielle et la bioproduction, ouvrant ainsi la voie à des avancées majeures.








