Command Palette
Search for a command to run...
Une Équipe De l'Université Chinoise De Hong Kong, De l'Université Du Zhejiang Et De l'Université Polytechnique De Macao a Proposé Un Cadre Général, Bi-TEAM, Pour Améliorer La Précision De La Prédiction Des Maladies Hémolytiques Par 350%, Intégrant La Sémantique Biologique Et La Précision chimique.

En biochimie et en ingénierie moléculaire, l'apprentissage par caractérisation s'impose progressivement comme une technologie clé pour élucider les fonctions moléculaires et accélérer la découverte de molécules thérapeutiques. La qualité des caractéristiques intégrées détermine souvent les performances maximales des tâches en aval, telles que la prédiction des propriétés des peptides et la conception de novo. En tant que molécule centrale reliant fonction biologique et propriétés chimiques, la modélisation de la structure et de la fonction des peptides revêt une importance considérable pour le développement de médicaments.Ces dernières années, l'introduction d'acides aminés non classiques a considérablement élargi l'espace fonctionnel des peptides et amélioré leur stabilité et leur biodisponibilité, mais les modifications chimiques complexes ont également posé de nouveaux défis aux méthodes de modélisation traditionnelles.Comment intégrer simultanément les informations sur l'évolution biologique et la rationalité chimique dans un modèle devient une question clé qui doit être traitée de toute urgence dans ce domaine.
Actuellement, la modélisation des peptides est principalement réalisée selon deux approches techniques.d'une part,Les modèles de langage protéique, tels que ESM et ProtT5, capturent le contexte biologique et les informations évolutives grâce à un pré-entraînement de séquences à grande échelle, fournissant des représentations biologiques transférables pour les tâches en aval.d'autre part,Pour résoudre le problème des modifications apportées aux acides aminés non classiques, les chercheurs ont utilisé un modèle de langage chimique pour capturer les détails chimiques grâce à une segmentation des mots au niveau atomique, compensant ainsi les lacunes des modèles de protéines au niveau chimique.
Cependant, les deux types de modèles présentent des limitations intrinsèques. Les modèles de langage protéique sont limités par l'ensemble naturel des caractères des acides aminés, ce qui rend difficile la prise en compte des résidus non classiques. Les méthodes existantes d'approximation ou d'extension du vocabulaire introduisent souvent des biais ou conduisent à une sparsité sémantique. Les modèles de langage chimique, quant à eux, ignorent le contexte biologique global, et la segmentation dense des mots dépasse facilement la fenêtre contextuelle, ce qui complique leur adaptation à la modélisation de longues séquences. Les modèles généraux souffrent également de biais de domaine.
Pour remédier aux problèmes susmentionnés, l'Université chinoise de Hong Kong, en collaboration avec l'Université polytechnique de Macao, l'Université du Zhejiang, le deuxième hôpital Xiangya de l'Université centrale du Sud et l'Université des sciences et technologies électroniques de Chine, a proposé un paradigme de modélisation de fusion sélective.Partant du principe que « la variation chimique est une perturbation locale de l’espace sémantique biologique », un cadre général, Bi-TEAM, a été conçu pour injecter une variation chimique locale dans le contexte protéique global.
Ce cadre utilise des représentations biologiques comme base sémantique et fusionne efficacement informations évolutives biologiques et raisonnement chimique grâce à l'injection adaptative de signaux chimiques. Dans de nombreuses tâches, Bi-TEAM surpasse systématiquement les modèles de référence les plus performants : après un partitionnement rigoureux des données basé sur la similarité squelettique, le coefficient de corrélation de Matthews est amélioré jusqu'à 661 TP3T ; dans la tâche de prédiction de l'hémolyse, la précision est améliorée de 3 501 TP3T.
Les résultats de recherche connexes, intitulés « Bi-TEAM : un cadre d'apprentissage de représentation inter-échelle unifié pour les biomolécules chimiquement modifiées », ont été publiés en tant que prépublication sur arXiv.
Points saillants de la recherche :
Le cadre Bi-TEAM peut intégrer de manière adaptative des propriétés biochimiques multi-échelles et servir de modèle a priori de haute fidélité pour une conception efficace des peptides.
* Les chercheurs ont évalué de manière exhaustive Bi-TEAM sur 10 ensembles de données divers dans 3 domaines biochimiques, atteignant des performances de pointe (SOTA) dans 7 tâches de prédiction clés.
* Ce modèle réalise une double percée dans les tâches de prédiction et de génération, améliorant le MCC de 66% sous une segmentation stricte de la similarité de l'échafaudage, tout en augmentant simultanément le taux de réussite de la conception de peptides cycliques pénétrant dans les cellules de près de 4 fois.

Adresse du document :
https://arxiv.org/abs/2603.01873
Suivez notre compte WeChat officiel et répondez « Bi-TEAM » en arrière-plan pour obtenir le PDF complet.
Une évaluation exhaustive a été menée, couvrant trois grands domaines biochimiques et 10 ensembles de données divers.
Cette étude évalue les propriétés selon deux dimensions : la prédiction des propriétés et la génération guidée, couvrant trois grands domaines de recherche : les peptides modifiés, les modifications post-traductionnelles (PTM) et les protéines naturelles, impliquant un total de 10 ensembles de données.
Dans le domaine des peptides modifiés, la recherche se concentre sur l'évaluation de la capacité du modèle à prédire la perméabilité membranaire.Les données d'entraînement principales proviennent de la base de données ProPAMPA, qui contient des cycles de 12 à 46 atomes, avec une distribution de longueur de séquence approximativement normale, mais présentant des queues de distribution importantes à ses extrémités. Elle contient également un grand nombre de résidus d'acides aminés naturels et non classiques, témoignant d'une grande diversité chimique. Après déduplication à l'aide de RDKit,Il contient un total de 6 876 séquences peptidiques cycliques non conjuguées.
Afin d'évaluer la capacité de généralisation du modèle, l'étude a introduit trois jeux de données expérimentales externes obtenus par voie humide : ProCacoPAMPA, CycPeptMPDB v1.2 et le jeu de données Rezai. Ces jeux de données couvrent des échantillons de peptides cycliques de différentes longueurs et de différents types structuraux. Plus précisément :
ProCacoPAMPA :Toutes les séquences de peptides cycliques transmembranaires de longueurs 6 et 10 ont été collectées à partir d'études existantes et intégrées dans un ensemble de données standardisé.
CycPeptMPDB v1.2 :La dernière version de la plus grande base de données publique sur la perméabilité membranaire des peptides cycliques non classiques, compilée à partir de 56 articles, contient 8 466 enregistrements. Dans cette étude, les chercheurs ont supprimé les échantillons dupliqués dans l’ensemble de données ProPAMPA, obtenant ainsi un sous-ensemble affiné de 1 230 points de données.
Rezai :Ces données comprennent la perméabilité membranaire passive de 11 peptides cycliques. Elles ont été obtenues par des expériences PAMPA et sont fréquemment utilisées pour la validation externe de modèles dans des conditions de faible envergure.
Afin de valider davantage la similarité du modèle avec un médicament et son association avec une maladie,Les chercheurs ont effectué une tâche de prédiction de la similarité avec un médicament sur l'ensemble de données PTM.Les données utilisées se répartissaient en deux catégories : les données relatives aux médicaments et les données associées aux maladies. Les premières étaient principalement constituées de longues séquences protéiques, avec des sites de modification présentant une distribution à longue traîne caractéristique. Les secondes provenaient essentiellement de bases de données telles que dbPTM et d’études d’association pangénomiques (GWAS). Bien que la distribution des sites de modification soit similaire à celle des premières, la gamme de longueurs de séquences était plus étendue, offrant ainsi un contexte structural plus diversifié.
Dans le domaine des protéines naturelles, les chercheurs se sont attachés à évaluer les performances du modèle en matière de prédiction de la solubilité et de l'hémolyse afin d'explorer les mécanismes clés de l'hémolyse des peptides et des variations de solubilité des protéines. Les jeux de données utilisés comprenaient principalement trois catégories : hémolyse, anticontamination et solubilité. Parmi eux :
Les données d'hémolyse proviennent de la base de données DBAASP v3.Il contient un total de 9 316 séquences composées d'acides aminés classiques de type L.
L'ensemble de données anti-pollution est principalement constitué de courtes séquences peptidiques.Les séquences présentent des longueurs comprises entre 5 et 10 résidus d'acides aminés, et leur distribution LogP est approximativement normale. Les échantillons présentent une bonne structure de regroupement dans l'espace des caractéristiques.
L'ensemble de données de solubilité est dérivé de séquences protéiques annotées avec PROSO II.Son étiquetage repose sur une analyse rétrospective de l'Initiative sur la structure des protéines.
Bi-TEAM : un cadre d’apprentissage unifié pour la caractérisation multi-échelle des biomolécules chimiquement modifiées
Bi-TEAM vise à relever le défi des modèles unimodaux existants en capturant simultanément des informations biologiques évolutives globales et des détails structuraux chimiques locaux (espace chimique à grain fin). Comme le montre la figure ci-dessous,Son idée centrale est de construire un système de représentation à double perspective qui intègre profondément l'espace biologique évolutif et l'espace de la structure chimique.Cela permet une modélisation plus précise des séquences peptidiques contenant des acides aminés non classiques.
En termes d'architecture globale,Le modèle utilise l'espace biologique construit par le modèle de langage protéique comme base sémantique, tirant pleinement parti des règles évolutives et des relations contextuelles apprises à partir de séquences naturelles à grande échelle.Parallèlement, un modèle de langage chimique (MLC) est introduit pour capturer les informations structurales à l'échelle atomique, palliant ainsi les limitations inhérentes au modèle de langage protéique (MLP) dans la gestion des modifications chimiques. Ces deux types de modèles se complètent au niveau de la représentation, élargissant conjointement le pouvoir expressif de la séquence d'entrée.

Lors du traitement de séquences peptidiques modifiées, Bi-TEAM les encode à l'aide de deux flux d'informations complémentaires :L'un d'eux est un flux de séquences biologiques.Les acides aminés modifiés sont associés aux acides aminés naturels structurellement les plus proches, évitant ainsi l'expansion de la table de segmentation des mots et préservant la sémantique évolutive qui peut être utilisée pour la modélisation.L'autre est le flux de représentation de type SELFIES.Il est utilisé pour décrire avec précision les changements de groupes fonctionnels et les structures de liaisons chimiques des résidus modifiés au niveau atomique, fournissant des informations structurelles stables pour les modèles de langage chimique.
Après avoir terminé l'encodage à double flux,Le modèle est fusionné à l'aide d'un mécanisme résiduel à double porte guidé par des indices d'embellissement sensibles à la position :En utilisant des représentations biologiques comme base sémantique, les signaux chimiques clés sont filtrés et injectés à l'aide d'unités de contrôle, tandis que les connexions résiduelles des caractéristiques biologiques sont préservées. Ceci permet au modèle d'établir une corrélation efficace entre les contraintes de séquence globales et les modifications chimiques locales, tout en maintenant la stabilité de l'apprentissage.
Au niveau de l'application, Bi-TEAM offre une bonne polyvalence.Lors du traitement de séquences protéiques naturelles non modifiées, le modèle peut directement omettre les étapes de cartographie et de localisation, s'adaptant aux tâches protéiques de routine sans ajuster l'architecture globale.
En matière de stratégies de formation,L'étude utilise un cadre en deux étapes de « pré-entraînement-ajustement fin » :Dans un premier temps, un pré-entraînement adaptatif au domaine a été réalisé sur deux types d'encodeurs de base, respectivement sur des séquences protéiques naturelles et des corpus de molécules chimiques de petite taille. Ensuite, grâce à un ajustement fin conjoint multitâche, le modèle a appris les règles de fusion des caractéristiques biologiques et chimiques dans différents scénarios de tâches, améliorant ainsi sa capacité de généralisation globale.
Bi-TEAM a réalisé une percée dans la conception de peptides cycliques pénétrants, augmentant le taux de réussite de 4,6 fois.
Afin de vérifier les capacités d'application de Bi-TEAM dans des espaces chimiques inconnus, cette étude se concentre sur la conception de peptides cycliques non canoniques pour le traitement de la dégénérescence maculaire néovasculaire liée à l'âge (DMLA néovasculaire) par voie intracellulaire, en utilisant l'administration non invasive de médicaments comme scénario. L'étude a systématiquement mené une expérience complète d'« analyse guidée par la prédiction » pour évaluer la performance du modèle dans la conception moléculaire guidée par les propriétés.
La DMLA néovasculaire est une cause majeure de cécité irréversible chez les personnes âgées. Sa principale pathologie est la néovascularisation choroïdienne et la fuite de VEGF induites par ce dernier. Actuellement, le traitement clinique repose principalement sur l'injection intravitréenne d'anti-VEGF de grande taille (comme l'aflibercept, 115 kDa).Cependant, ces médicaments ont du mal à pénétrer la barrière physiologique de l'œil, et les injections à long terme peuvent entraîner des complications et des problèmes d'observance.La conception de peptides capables de se lier spécifiquement à l'aflibercept et de favoriser son transport transmembranaire ouvrirait de nouvelles perspectives pour les collyres non invasifs. Comparés aux peptides linéaires facilement dégradables et à courte demi-vie, les peptides cycliques, grâce à leur structure plus stable et leur perméabilité accrue, sont considérés comme des vecteurs plus adaptés, ce qui motive principalement les chercheurs à se concentrer sur leur conception.
Cette étude a d'abord effectué une prédiction et une évaluation des peptides pénétrant dans les cellules (PPC) afin de fournir une base pour les tâches de génération ultérieure.L'ensemble de données a été construit selon le schéma standard pLM4CPP, intégrant des bases de données telles que CPPsite2.0, C2Pred et CellPPD. Après criblage et déduplication, 1 399 échantillons positifs (peptides pénétrants validés expérimentalement) et 4 080 échantillons négatifs ont été obtenus. Les modèles de comparaison comprenaient des modèles d'intégration de protéines courants tels que SeqVec, ESM2 et ProtT5, et les métriques d'évaluation incluaient l'ACC, le BACC, la sensibilité (Sn), la spécificité (Sp), le coefficient de corrélation de Matthews (MCC) et l'aire sous la courbe ROC (AUC).
Les résultats montrent que Bi-TEAM a obtenu les meilleures performances pour tous les indicateurs :L'ACC a été améliorée de 5,521 TP3T par rapport à SeqVec, la BACC de 5,881 TP3T par rapport à ESM2-480, la sensibilité de 12,581 TP3T, la spécificité de 1,451 TP3T par rapport à ProtT5-XL BFD, le coefficient de corrélation de Matthews (MCC) de 14,681 TP3T par rapport à SeqVec et l'aire sous la courbe (AUC) de 8,451 TP3T par rapport à ESM2-480. Ces améliorations significatives de la sensibilité et du MCC indiquent que le modèle présente un net avantage pour l'identification des peptides pénétrants.
S’appuyant sur ces bases, des recherches supplémentaires seront menées sur des expériences de génération de peptides cycliques guidées par les propriétés.En utilisant BoltzDesign1 comme cadre de base, 1 000 peptides cycliques de longueur 10-20 ont été générés dans deux conditions : l'une utilisant uniquement les contraintes structurelles par défaut, et l'autre introduisant Bi-TEAM comme guide de gradient supplémentaire pendant le processus de génération.
Le critère de succès était un rapport de cotes logarithmique de prédiction Bi-TEAM supérieur à 0,5. Les résultats ont montré que…Les méthodes traditionnelles ont donné un taux de réussite de seulement 6,71 TP3T dans la génération de peptides cycliques pénétrant dans les cellules, tandis que les méthodes guidées par Bi-TEAM ont amélioré ce taux à 30,71 TP3T.Parallèlement, la qualité structurelle n'a pas diminué : le pLDDT moyen du complexe peptide-Aflibercept généré a dépassé 0,82, indiquant que le modèle a maintenu une bonne confiance structurelle et une bonne stabilité de l'interface de liaison tout en améliorant la pénétration.

Pour comprendre le mécanisme de guidage, les chercheurs ont analysé plus en détail les motifs de résidus de la séquence générée. Des études antérieures ont montré que le triplet hydrophobe composé de tryptophane (W), de phénylalanine (F) et de tyrosine (Y), ainsi que les résidus chargés positivement tels que l'arginine (R) et la lysine (K), sont des caractéristiques clés permettant aux peptides pénétrant dans les cellules de traverser la membrane.
L'analyse a révélé que,Guidée par Bi-TEAM, la fréquence de cooccurrence du triplet hydrophobe et de deux résidus chargés positivement dans la séquence générée a été considérablement augmentée, et la distribution du nombre de résidus a également montré une tendance cohérente.Ce profil d'enrichissement est parfaitement cohérent avec les règles structure-fonction connues des peptides pénétrants, indiquant que Bi-TEAM peut non seulement capturer les mécanismes biologiques pertinents, mais aussi augmenter significativement la probabilité de séquences possédant des propriétés de pénétration membranaire lors du processus de génération. L'analyse des variables de contrôle a par ailleurs exclu l'influence de la longueur du peptide (10 à 20 résidus), démontrant que le modèle oriente effectivement la distribution de l'échantillonnage vers un espace de colocalisation chimio-biologique plus favorable au transport membranaire.
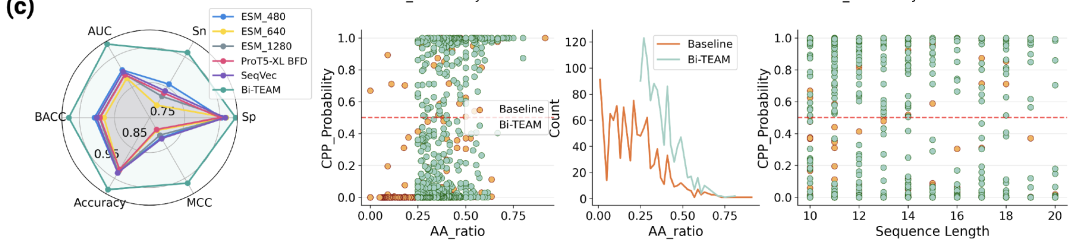
Dans la partie centrale : la relation entre l’abondance des résidus hydrophobes clés et la probabilité de pénétration ;
À droite : Relation entre la longueur du peptide cyclique et la probabilité de pénétration
Enfin, l'étude a validé les résultats au niveau structural par le biais d'une étude de cas. Les chercheurs ont d'abord visualisé la structure tridimensionnelle du dimère d'aflibercept et coloré sa surface moléculaire en fonction du potentiel électrostatique. Ils ont ensuite utilisé AlphaFold3 pour prédire et concevoir la structure du complexe formé par le peptide cyclique et l'aflibercept. L'analyse a identifié deux poches de liaison potentielles pour le peptide cyclique : une cavité hydrophobe composée de trois anneaux et une autre formée par une structure cyclique et un fragment de feuillet β. Ces informations structurales constituent une base importante pour l'optimisation ultérieure du peptide cyclique et ses applications cliniques potentielles.

Se concentrer sur l'innovation technologique dans le domaine du développement des médicaments peptidiques
Dans le domaine de la science des peptides, de la recherche fondamentale à la translation clinique, de nombreux instituts de recherche à travers le monde explorent activement de nouvelles voies technologiques et options de traitement pour vaincre les maladies majeures.
Par exemple, l'équipe de biologie structurale de l'École de biochimie de l'Université de Bristol, au Royaume-Uni.Des techniques avancées telles que la cryo-microscopie électronique et la cristallographie aux rayons X sont utilisées pour analyser la structure fine du système immunitaire, et c'est sur cette base que la conception de médicaments peptidiques est réalisée.Ils tentent de développer des candidats médicaments de nouvelle génération pour le traitement des maladies auto-immunes en concevant des molécules peptidiques cycliques capables d'activer précisément le système du complément humain.
Parallèlement, le projet ToxiCode, une collaboration entre le King's College de Londres et l'Université de Zagreb, explore une voie unique pour découvrir de nouveaux médicaments à partir de venins animaux.Ce projet combine intelligence artificielle et biologie synthétique, utilisant un système d'IA hybride pour apprendre les modèles de séquences peptidiques et leurs relations structure-activité, permettant la conception rapide de nouveaux peptides bioactifs ciblant le cancer, les troubles neurologiques et les maladies infectieuses.Cela offre un nouveau cadre méthodologique pour une découverte de médicaments durable et éthique.
Ceci démontre que le développement de médicaments peptidiques instaure progressivement un nouveau paradigme de recherche : la biologie structurale, l’intelligence artificielle et la biologie chimique convergent de plus en plus, et les frontières entre recherche fondamentale et développement industriel s’estompent. De nouvelles molécules émergent souvent de combinaisons technologiques interdisciplinaires, mais leur application clinique dépend véritablement du processus de translation qui s’établit progressivement, de la découverte en laboratoire à l’industrialisation. Dans ce contexte, les molécules peptidiques, grâce à leurs propriétés uniques à mi-chemin entre les petites et les grandes molécules, sont réévaluées et trouvent de nouvelles applications dans un nombre croissant de domaines thérapeutiques.
Liens de référence :
1.https://www.bristol.ac.uk/news/2025/november/bristol-researcher-awarded-over-850000-to-develop-new-treatments.html
2.https://www.kcl.ac.uk/news/kings-to-collaborate-in-venom-based-drug-discovery-project
3.https://mp.weixin.qq.com/s/X67D1qrUzclwOsJ9cKUtZg








