Command Palette
Search for a command to run...
Sélectionné Pour NeurIPS 2025, l'Université De Toronto Et d'autres Ont Proposé Un Cadre Ctrl-DNA Pour Obtenir Un « Contrôle Ciblé » De l'expression Des Gènes Dans Des Cellules spécifiques.

La régulation précise de l'expression des gènes dans des cellules spécifiques est essentielle aux progrès dans des domaines tels que la thérapie génique et la biologie synthétique. Ce processus repose sur une classe de séquences d'ADN appelées « éléments cis-régulateurs » (ECR), tels que les promoteurs et les activateurs. Ces derniers agissent comme des « interrupteurs » pour les gènes, déterminant leur activation ou leur désactivation dans les cellules cibles, tout en évitant toute activation anormale dans d'autres cellules normales. Cependant,Le nombre de CRE efficaces naturellement présents est limité et difficile à adapter avec précision à divers scénarios d’application biomédicale.Plus important encore, les possibilités offertes par les séquences d'ADN connaissent une croissance exponentielle. Par exemple, une séquence de 100 bases possède 4¹⁰⁰ combinaisons. Il est extrêmement difficile de les vérifier une par une par des expériences. Cette méthode est non seulement chronophage et laborieuse, mais elle ne répond pas non plus aux besoins pratiques.
Les méthodes actuelles basées sur l’apprentissage profond ont considérablement amélioré l’efficacité expérimentale, mais les méthodes existantes sont encore confrontées à de multiples défis.Par exemple, certaines méthodes s'appuient sur des mutations dans l'ADN existant ou sur l'optimisation aléatoire des séquences, ce qui peut facilement tomber dans le piège de l'« optimalité locale », entraînant une diversité insuffisante des séquences efficaces générées. Si les approches basées sur des modèles de langage autorégressifs peuvent capturer des schémas de séquences d'ADN, elles ne peuvent qu'« imiter des séquences connues » et sont incapables d'explorer de nouveaux CRE spécifiques aux cellules. Si les méthodes d'apprentissage par renforcement (RL) améliorent les effets régulateurs dans les cellules cibles, elles négligent le contrôle des « effets secondaires » sur les autres cellules. De plus, ces cadres de conception standard négligent souvent les considérations de plausibilité biologique. Les séquences générées peuvent ne pas correspondre aux sites clés de liaison aux facteurs de transcription (TFBS), entraînant ainsi l'échec des fonctions régulatrices réelles.
Pour combler le vide dans la conception précise du CRE spécifique aux cellules, une équipe de l’Université de Toronto, en collaboration avec le laboratoire Changping et d’autres institutions, a développé un cadre d’apprentissage par renforcement contraint appelé Ctrl-DNA.Ce cadre, basé sur un modèle de langage ADN pré-entraîné, utilise un algorithme d'apprentissage par renforcement pour atteindre simultanément un double objectif lors du processus d'optimisation : maximiser l'activité régulatrice des CRE dans les cellules cibles tout en limitant strictement leur activité dans les cellules non ciblées. De plus, l'outil mathématique des multiplicateurs de Lagrange est utilisé pour équilibrer ces deux exigences, et la distribution des TFBS dans l'ADN réel est référencée afin de garantir la validité biologique des séquences générées.
Les résultats de l’étude ont montré queDans les tâches de conception de 6 cellules humaines, le CRE généré par Ctrl-DNA a considérablement surpassé les méthodes existantes dans deux indicateurs clés : « activité élevée dans les types de cellules cibles » et « contrainte dans les types de cellules non cibles ».Elle conserve également une diversité significative, offrant de nouvelles solutions pour la biologie synthétique afin de « créer des systèmes contrôlables », la thérapie génique pour « éviter les risques hors cible » et la médecine de précision pour « réaliser une personnalisation au niveau cellulaire ».
Les résultats de recherche pertinents ont été publiés sur la plateforme de pré-impression arXiv sous le titre « Ctrl-DNA : Constrained Reinforcement Learning for Cell-Specific Cis-Regulatory Element Design » et ont été sélectionnés pour NeurIPS 2025.
Points saillants de la recherche :
* Un nouveau cadre d’apprentissage par renforcement prenant en compte les contraintes est proposé pour fournir des outils permettant de concevoir des CRE pour une expression génétique précise et spécifique au type de cellule.
* Simplification du processus d'optimisation, amélioration de l'efficacité expérimentale et réduction des coûts de calcul
* Des expériences ont vérifié que Ctrl-ADN a à la fois une efficacité fonctionnelle et une plausibilité biologique

Adresse du document :
https://arxiv.org/abs/2505.20578
Suivez le compte officiel et répondez « Ctrl-DNA » pour obtenir le PDF complet
Autres articles sur les frontières de l'IA :
Ensemble de données : basé sur de véritables ensembles de données de promoteurs et d'amplificateurs humains
Dans cette étude, les chercheurs ont utilisé de véritables ensembles de données de promoteurs et d’amplificateurs humains pour évaluer et valider Ctrl-DNA.
dans,L'ensemble de données sur les promoteurs humains contient des données sur l'activité des promoteurs de trois lignées cellulaires dérivées de la leucémie.Les trois lignées cellulaires sont : Jurkat, K562 et THP1. Toutes trois sont des lignées cellulaires hématopoïétiques dérivées du mésoderme et présentent une forte similarité biologique. Chaque séquence de cet ensemble de données comporte 250 paires de bases. Voir le tableau ci-dessous.
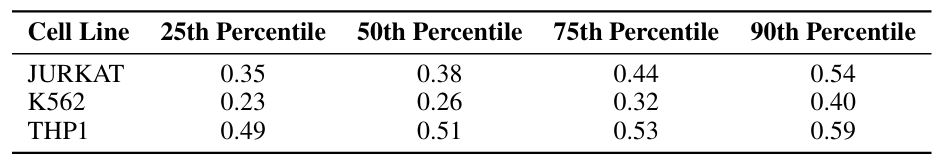
L'ensemble de données Human Enhancer contient des données d'activité CRE provenant de trois lignées cellulaires mesurées par un test de rapporteur massivement parallèle (MPRA).Les trois lignées cellulaires sont : HepG2 (lignée hépatique), K562 (lignée érythroïde) et SK-N-SH (lignée neuroblastome). Chaque séquence de cet ensemble de données comporte 200 paires de bases. Comme l'indique le tableau suivant :
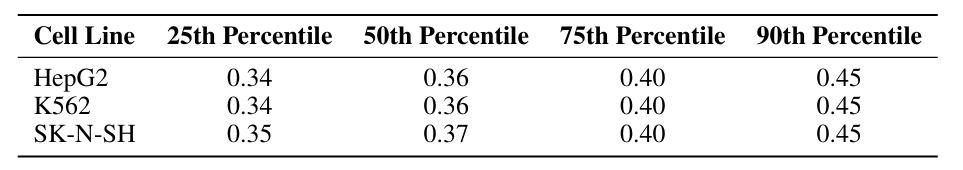
Il est à noter que dans la lignée cellulaire THP1, l'activité du 25e percentile a atteint 0,49, ce qui montre une distribution biaisée vers la droite. Ce biais de distribution pourrait expliquer en partie la difficulté accrue à limiter l'activité dans la lignée cellulaire THP1.
Architecture du modèle : Basée sur un modèle de langage ADN pré-entraîné, combiné à une relaxation lagrangienne
Ctrl-DNA est un cadre de conception de séquences d'ADN régulatrices basé sur l'apprentissage par renforcement contraint, dont l'objectif principal est de générer des CRE avec une spécificité de type cellulaire contrôlable.En termes de mise en œuvre fonctionnelle, il faut maximiser la fitness des CRE dans les cellules cibles, c'est-à-dire améliorer l'expression génique, tout en contrôlant rigoureusement la fitness dans les cellules hors cible, dans les limites d'un seuil prédéfini. Parallèlement, il faut garantir que les séquences générées sont conformes aux lois biologiques réelles afin d'éviter que les résultats expérimentaux soient satisfaisants, mais que l'application soit inefficace.
À cette fin, les chercheurs ont pris en compte la facilité d’utilisation, la rationalité et d’autres aspects du cadre, et ont réalisé une conception détaillée du cadre, comme le montre la figure suivante :
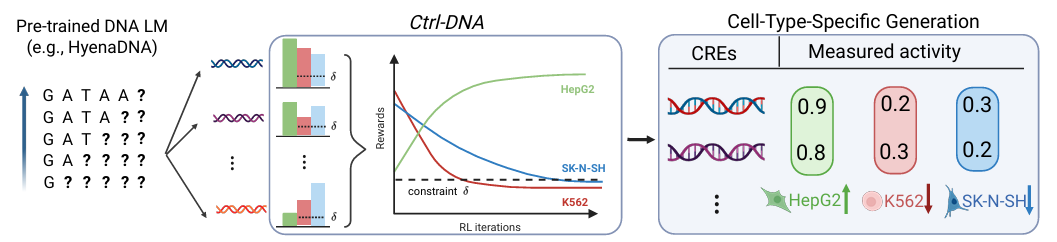
En termes de modèles et d’entrées,Ctrl-DNA affine le modèle de langage génomique autorégressif HyenaDNA pré-entraîné sur le génome humain comme modèle de politique initial, et utilise l'architecture Enformer pour former le modèle de récompense spécifique au type de cellule.Combinées aux données de « séquence-fitness » mesurées par des expériences de rapports massivement parallèles, les récompenses des cellules cibles et les récompenses des cellules hors cible sont calculées séparément.
Au niveau de la modélisation des problèmes,Les chercheurs ont transformé la conception de séquences d’ADN en un processus de décision de Markov contraint (CMDP). Le mécanisme d'optimisation principal de Ctrl-DNA utilise l'optimisation par politique relative par lots sous contrainte (CBROP). Ce mécanisme transforme un problème d'optimisation sous contrainte en un problème d'optimisation primal-dual sans contrainte grâce à la relaxation lagrangienne. Le processus d'optimisation est itératif, les mises à jour de politique suivant le gradient de la fonction objective lagrangienne au rythme d'apprentissage. Les récompenses des cellules hors cible sont limitées par l'ajustement du multiplicateur lagrangien : il augmente le multiplicateur lagrangien pour renforcer la contrainte pour les cellules hors cible dépassant un seuil, et il diminue le multiplicateur lagrangien pour l'affaiblir pour les cellules hors cible atteignant ce seuil.
Afin de réduire la complexité de la formation, Ctrl-DNA abandonne le recours aux modèles de valeur dans l’apprentissage par renforcement traditionnel.L'avantage normalisé est calculé directement sur la base des statistiques de données par lots pour guider l'optimisation de la stratégie afin de sélectionner des séquences avec « une récompense cible élevée + une récompense hors cible faible ».
Pour concevoir la fonction objective de mise à jour de la stratégie, les chercheurs ont adopté une combinaison d'objectifs de remplacement par élagage et de régularisation KL. Grâce à l'élagage, ils ont limité les mutations de stratégie et introduit la divergence KL entre la stratégie actuelle et la stratégie de référence initiale afin de garantir la cohérence de la séquence générée avec le profil d'ADN naturel, formant ainsi une fonction objective de mise à jour de stratégie.
Pour garantir une plausibilité biologique accrue, Ctrl-DNA introduit la corrélation de fréquence TFBS comme contrainte supplémentaire. Dans un premier temps, les TFBS sont scannés à l'aide de l'outil FIMO à partir de séquences CRE réelles et hautement spécifiques afin de construire un véritable vecteur de fréquence TFBS. Le vecteur de fréquence TFBS correspondant est ensuite calculé pour chaque séquence générée. Le coefficient de corrélation de Pearson est ensuite utilisé comme contrainte supplémentaire, tandis que le multiplicateur de Lagrange correspondant est limité à [0, λmax] (λmax ≤ 1). Cela équilibre la plausibilité biologique et l'optimisation objective, évitant ainsi les contraintes excessives susceptibles de réduire les capacités d'exploration du modèle.
Afin de garantir la stabilité de l'entraînement du modèle, les chercheurs ont démontré les paramètres d'hyperparamètres utilisés dans les expériences. Tous les modèles ont été entraînés avec l'optimiseur Adam, avec un taux d'apprentissage des politiques de 1e-4, une taille de lot de 256 et 100 périodes d'entraînement. Les expériences ont été entraînées sur un seul GPU NVIDIA A100 doté de 40 Go de mémoire, comme illustré dans la figure ci-dessous.
Résultats expérimentaux : Comparé à 8 types de méthodes de base, Ctrl-DNA présente des avantages évidents
L'expérience d'évaluation des performances de Ctrl-DNA s'articule autour de deux tâches de conception majeures : les activateurs et les promoteurs humains, couvrant les six lignées cellulaires mentionnées ci-dessus. Elle est comparée à huit types de méthodes de référence, dont des algorithmes évolutionnaires (dont AdaLead, l'optimisation bayésienne (BO), CMA-ES, PEX), des modèles génératifs (RegLM) et des méthodes d'apprentissage par renforcement (dont TACO, PPO et PPO-Lagrangien), afin de vérifier son efficacité et sa faisabilité à partir de multiples dimensions telles que la spécificité du type cellulaire, la plausibilité biologique et la diversité des séquences.
Ctrl-DNA présente des avantages significatifs en termes de confinement spécifique au type de cellule.Comme le montre la figure ci-dessous, l'axe horizontal représente la fitness des types de cellules hors cible, tandis que l'axe vertical représente la fitness des types de cellules cibles. La méthode illustrée en haut à droite représente le meilleur équilibre entre maximisation de la fitness des cellules cibles et minimisation de l'expression hors cible.
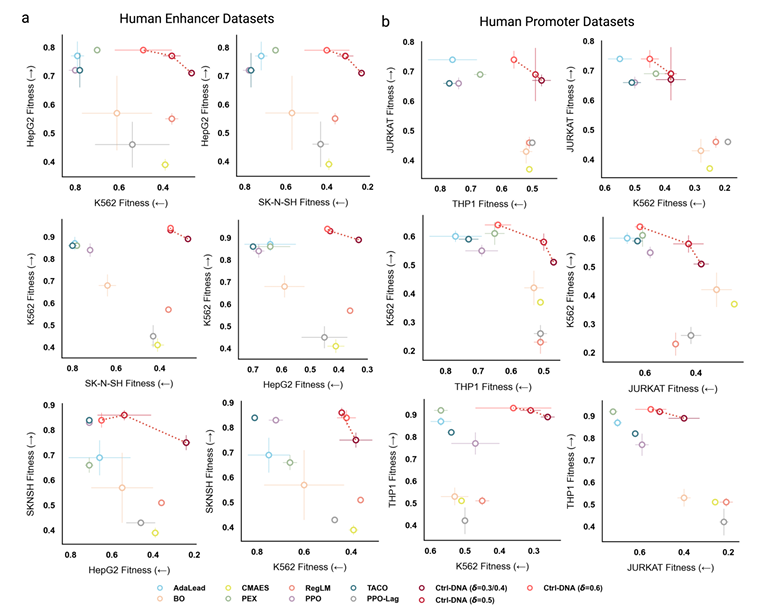
Pour la conception d'amplificateurs, Ctrl-DNA a systématiquement atteint la meilleure aptitude cellulaire cible tout en respectant les contraintes hors cible à tous les seuils de contrainte (δ = 0,3, 0,5 et 0,6). Cela signifie qu'il a maximisé l'aptitude cellulaire cible tout en respectant strictement les contraintes hors cible. De plus, si des méthodes telles que TACO et CMAES ont permis d'obtenir une expression élevée dans les cellules cibles, elles n'ont pas réussi à supprimer l'aptitude cellulaire hors cible, ce qui a entraîné une faible spécificité cellulaire.
Pour la conception du promoteur, les trois types de cellules cibles étant des cellules hématopoïétiques d'origine mésodermique, ils présentent de grandes similitudes transcriptionnelles, ce qui représente un défi de taille. Cependant, Ctrl-DNA s'avère néanmoins performant. L'expérience a défini trois seuils de contrainte différents (δ = 0,4, 0,5 et 0,6). Ctrl-DNA a surpassé toutes les valeurs de référence en maximisant la fitness du type de cellule cible et en respectant les seuils de contrainte δ = 0,5 et 0,6. Il convient également de noter que pour des cas tels que les cellules THP1 où la distribution d'activité est asymétrique à droite (comme mentionné dans la section sur l'ensemble de données ci-dessus, l'activité du 25e percentile a atteint 0,49), aucune méthode ne peut supprimer son activité hors cible au seuil strict de δ = 0,4, mais Ctrl-DNA est la méthode qui se rapproche le plus de l'exigence de contrainte parmi toutes les méthodes.
Lors de la validation de la plausibilité biologique, comme le montre la figure ci-dessous, Ctrl-DNA a obtenu la différence de récompense (ΔR) la plus élevée parmi tous les types cellulaires pour les promoteurs et activateurs humains, indiquant qu'il optimise mieux la fitness spécifique des séquences d'ADN. Concernant la pertinence des motifs, Ctrl-DNA a également obtenu de meilleures performances dans la plupart des types cellulaires, à l'exception du promoteur THP1.
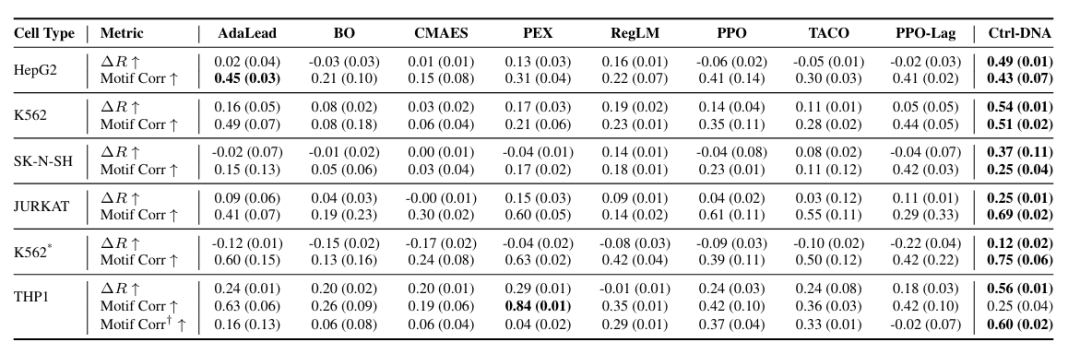
Pour explorer plus en détail cette divergence, les chercheurs ont extrait des motifs des séquences promotrices au 90e percentile de la fitness de THP1. En utilisant un seuil de q < 0,05 pour éviter les faux positifs, ils ont réévalué la corrélation des motifs entre les séquences générées et l'ensemble de référence, représentée par le motif Corr† dans la figure ci-dessus. Les résultats ont montré que Ctrl-DNA surpassait toutes les valeurs de référence, même dans ce contexte rigoureux, son coefficient de corrélation augmentant à 0,60, tandis que les corrélations pour la plupart des valeurs de référence diminuaient, démontrant sa capacité à capturer préférentiellement les motifs régulateurs fonctionnellement significatifs.
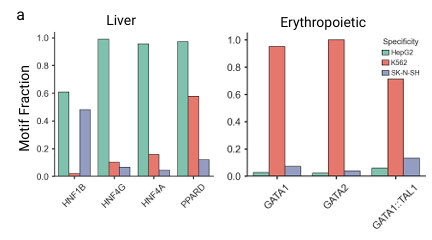
Pour analyser plus en détail la fréquence des TFBS spécifiques trouvés, les chercheurs ont examiné spécifiquement les séquences générées pour les motifs spécifiques à la lignée cellulaire d'hépatocytes HepG2 et à la lignée cellulaire érythroïde K562.Comme le montre la figure ci-dessous, la séquence HepG2 générée par Ctrl-DNA présente la plus forte fréquence de motifs spécifiques au foie, tels que HNF4A et HNF4G. De même, la séquence générée pour K562 présente la plus forte fréquence de motifs spécifiques aux érythroïdes, tels que GATA1 et GATA2. Cela démontre que Ctrl-DNA optimise non seulement la capacité de la cellule cible, mais apprend également des schémas de régulation qui reflètent la spécificité du type cellulaire sous-jacent.
En termes de diversité de séquence, Ctrl-DNA a atteint une diversité comparable ou supérieure à la plupart des lignes de base, confirmant sa capacité à générer des séquences diverses sans sacrifier le contrôle réglementaire.Comme le montre la figure suivante :
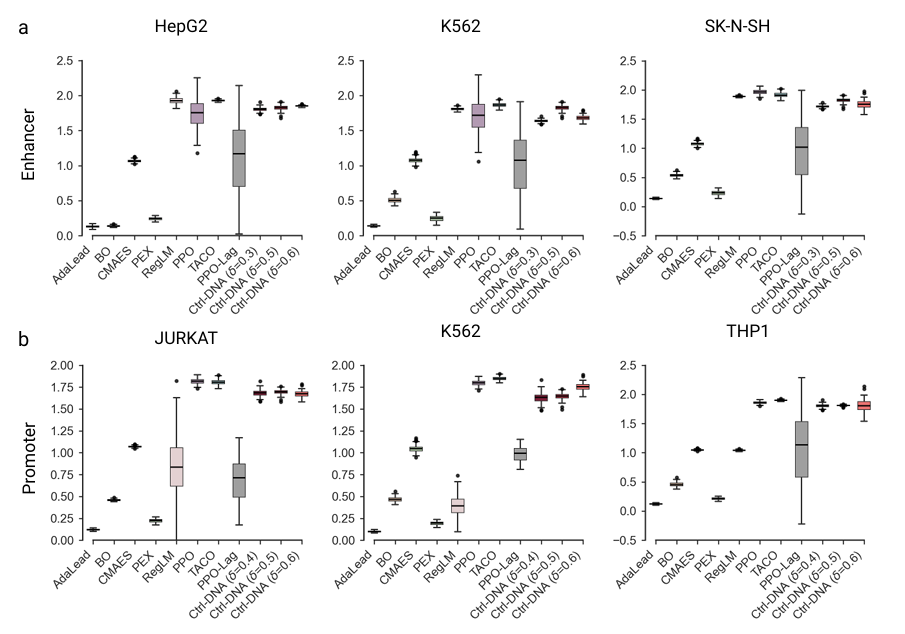
Enfin, les chercheurs ont validé l'efficacité du module central Ctrl-DNA par des expériences d'ablation. Le rôle du module de régularisation TFBS a également été confirmé, guidant efficacement les séquences vers des schémas biologiquement réalistes.
La conception d'un « commutateur » d'ADN piloté par l'IA ouvre un nouveau chapitre
Dans le passé, la conception de « commutateurs » de séquences d’ADN régulatrices reposait principalement sur des « essais et erreurs » au travers d’un grand nombre de criblages manuels répétés.Désormais, grâce à la combinaison de la technologie de l'IA, nous pouvons utiliser des algorithmes pour prédire « quelles séquences d'ADN ont la plus grande correspondance avec la protéine régulatrice cible », ce qui améliore considérablement l'efficacité et la précision de la conception.C'est également la raison principale pour laquelle la conception de commutateurs d'ADN pilotés par l'IA est devenue une nouvelle direction, qui à son tour favorise directement des domaines tels que la thérapie génique et la biologie synthétique de « étendu » à « précis ».
Cet article n'est qu'un des fruits de la conception de commutateurs d'ADN pilotés par l'IA. De nombreux laboratoires ont déjà mené des recherches sur ce sujet.
Par exemple, une équipe du Jackson Laboratory, du Broad Institute et de l'Université Yale a publié une étude dans Nature intitulée « Conception guidée par machine d'éléments cis-régulateurs ciblant le type de cellule ».L’étude a utilisé l’intelligence artificielle pour concevoir des milliers de nouveaux commutateurs ADN.Ces commutateurs permettent de contrôler précisément l'expression des gènes dans différents types de cellules. Plus précisément, les chercheurs ont construit un réseau neuronal convolutionnel profond (Malinois) capable de prédire avec précision l'activité des CRE et ont développé une plateforme modulaire (CODA) pour concevoir des CRE dotés de fonctions spécifiques. Cette plateforme offre des outils puissants pour le développement de gènes rapporteurs, la thérapie CRISPR, les méthodes de remplacement génique, et bien plus encore.
Adresse papier:
https://www.nature.com/articles/s41586-024-08070-z
De plus, RegLM, mentionné dans l'article ci-dessus, provient de Genentec. Dans l'étude intitulée « Concevoir un ADN régulateur réaliste avec des modèles de langage autorégressifs »,Nous introduisons un cadre appelé RegLM, qui est basé sur un modèle de langage autorégressif combiné à un modèle de séquence-fonction supervisé pour concevoir des CRE synthétiques avec des propriétés spécifiques.De même, RegLM s'appuie sur le framework HyenaDNA. Il code les étiquettes fonctionnelles sous forme de jetons d'indice et les ajoute au préfixe de la séquence d'ADN. Il entraîne ou affine le modèle pour prédire le jeton suivant et génère ainsi des séquences d'ADN possédant les fonctions souhaitées. Parallèlement, il combine un modèle de régression séquence-activité supervisé pour cribler les séquences générées.
Adresse du document :
https://genome.cshlp.org/content/34/9/1411.full#aff-1
En résumé, le développement de Ctrl-DNA constitue sans aucun doute une nouvelle avancée dans la conception de commutateurs d'ADN. Bien que certains problèmes ou domaines nécessitent encore des améliorations urgentes, comme l'intégration de contraintes biologiques supplémentaires pour améliorer la rationalité et la fonctionnalité de la séquence générée, et que l'ajustement du multiplicateur de Lagrange reste une question d'expérience, le développement et l'amélioration de ces outils ont indéniablement ouvert un nouveau chapitre pour la conception de commutateurs d'ADN, tout en favorisant le développement continu de la science interdisciplinaire de l'intelligence artificielle et de la biologie.








