Command Palette
Search for a command to run...
Relever Le Défi De La Modélisation À l'échelle Atomique De l'hétérogénéité Conformationnelle Des Protéines ! Analyse Du Cadre PLACER Par l'équipe De David Baker

Dans le monde moléculaire, les interactions entre les protéines et les acides nucléiques, les petites molécules organiques et inorganiques, et les ions métalliques sont essentielles aux fonctions vitales. Chaque reconnaissance et liaison de ces interactions peut affecter les fonctions biologiques, déterminer l'efficacité des médicaments, et même influencer le succès ou l'échec de la conception de nouvelles enzymes. Cependant, la modélisation à l'échelle atomique de ces interactions et de leur hétérogénéité conformationnelle demeure un défi de taille pour l'industrie.
Les outils d'amarrage de petites molécules basés sur l'apprentissage profond (DL), tels que DiffDock, ont amélioré la précision par rapport aux méthodes précédentes.Cependant, la différence de performance n'est pas significative pour les tâches de haute précision.De plus, les performances se dégradent considérablement lors de la rencontre de récepteurs inconnus ; en outre, diverses méthodes basées sur l'apprentissage profond ont été développées pour générer des conformations de petites molécules à partir de structures chimiques.Cependant, ces méthodes ne modélisent généralement que des catégories spécifiques d'objets interagissant, limitant ainsi leur capacité à caractériser tout le spectre des fonctions protéiques.
Sur cette base, une équipe de recherche dirigée par le professeur David Baker, lauréat du prix Nobel et professeur à l'Université de Washington, a développé un réseau neuronal graphique appelé PLACER (Protein-Ligand Atomistic Conformational Ensemble Resolver).Il peut générer avec précision les structures de diverses petites molécules organiques à partir de la composition atomique et des informations de liaison des petites molécules ; et, compte tenu de l'environnement structurel macroscopique des protéines, il peut construire les structures détaillées des petites molécules et des chaînes latérales des protéines pour les tâches d'amarrage protéine-petite molécule.Dans le cadre de recherches sur la conception d'enzymes, cette équipe a découvert que l'utilisation de PLACER pour évaluer la précision des sites actifs conçus et le degré de préorganisation permet d'améliorer significativement le taux de réussite de la conception et l'activité enzymatique. Par exemple, les chercheurs ont obtenu une antialdolase pré-organisée avec un kcat/KM de 11 000 M⁻¹·min⁻¹.Il surpasse de loin tous les résultats de conception antérieurs à l'avènement de l'apprentissage profond.
Les résultats de recherche connexes, intitulés « Modélisation des ensembles conformationnels protéine-petite molécule avec PLACER », ont été publiés dans les Actes de l'Académie nationale des sciences (PNAS).
Points saillants de la recherche :
* PLACER est rapide et aléatoire, ce qui lui permet de générer rapidement un grand nombre d'échantillons de prédiction pour représenter la distribution de l'hétérogénéité conformationnelle.
* En utilisant une représentation unifiée au niveau atomique pour toutes les interactions, PLACER peut être facilement étendu au-delà des biomolécules, telles que les molécules macrocycliques et d'autres petites molécules complexes.
* PLACER est d'une grande valeur pour la conception informatique d'enzymes et la conception de conjugués de petites molécules : il peut rapidement évaluer la précision de la reconstruction du site actif conçu et la pré-organisation des principaux groupes fonctionnels de chaînes latérales catalytiques/interagissantes.

Adresse du document :
https://www.biorxiv.org/content/10.1101/2024.09.25.614868v2
Suivez notre compte WeChat officiel et répondez « conception enzymatique » en arrière-plan pour obtenir le PDF complet.
Jeux de données : La construction de données multiniveaux et diversifiées confirme une excellente capacité de généralisation
Pour la prédiction de la conformation des petites molécules, l'équipe a sélectionné plus de 226 000 structures cristallines de petites molécules organiques non polymères issues de la Cambridge Structural Database (CSD) comme ensemble d'entraînement et 7 116 échantillons comme ensemble de validation. Chaque molécule fournit des informations complètes sur sa composition atomique et ses liaisons chimiques, tandis que les coordonnées atomiques sont initialisées aléatoirement, ce qui permet au modèle d'apprendre à reconstituer des structures précises même en présence de bruit.Cette stratégie d'entraînement garantit non seulement que le modèle puisse saisir les changements subtils des petites molécules dans différentes conformations, mais permet également la génération d'un ensemble diversifié de conformations moléculaires grâce à de multiples exécutions.
Concernant les systèmes protéine-petite molécule, l'équipe de recherche a sélectionné des structures à haute résolution (< 2,5 Å) issues de la Protein Data Bank (PDB), incluant des complexes protéine-petite molécule, pour un total d'environ 113 000 échantillons d'entraînement et 7 090 échantillons de validation. À noter,L'équipe a exclu uniquement les molécules d'eau, mais a conservé les informations sur les petites molécules potentiellement non biologiques (telles que les solvants) car elles fournissaient encore des indices précieux sur les préférences physico-chimiques des interfaces moléculaires.Les données d'entraînement sont tronquées pour contenir un maximum de 600 atomes lourds et perturbées par un bruit gaussien autour des centres atomiques sélectionnés aléatoirement afin de simuler l'environnement dynamique complexe des protéines et des petites molécules dans la réalité.
Cette structure de données multicouche et diversifiée garantit que PLACER présente une excellente capacité de généralisation lors du traitement de tout, des petites molécules uniques aux systèmes complexes protéine-petite molécule.
Le réseau neuronal PLACER utilise une architecture à trois voies, axée sur les chaînes latérales au niveau atomique et les conformations des petites molécules.
PLACER est un réseau neuronal de débruitage qui prend en entrée des structures protéiques partiellement perturbées et des informations sur la structure chimique (à l'exclusion des coordonnées) des molécules interagissantes. Il fournit en sortie la structure atomique complète du complexe et l'incertitude de la position de chaque atome dans le modèle de prédiction, comme illustré dans la figure A ci-dessous.
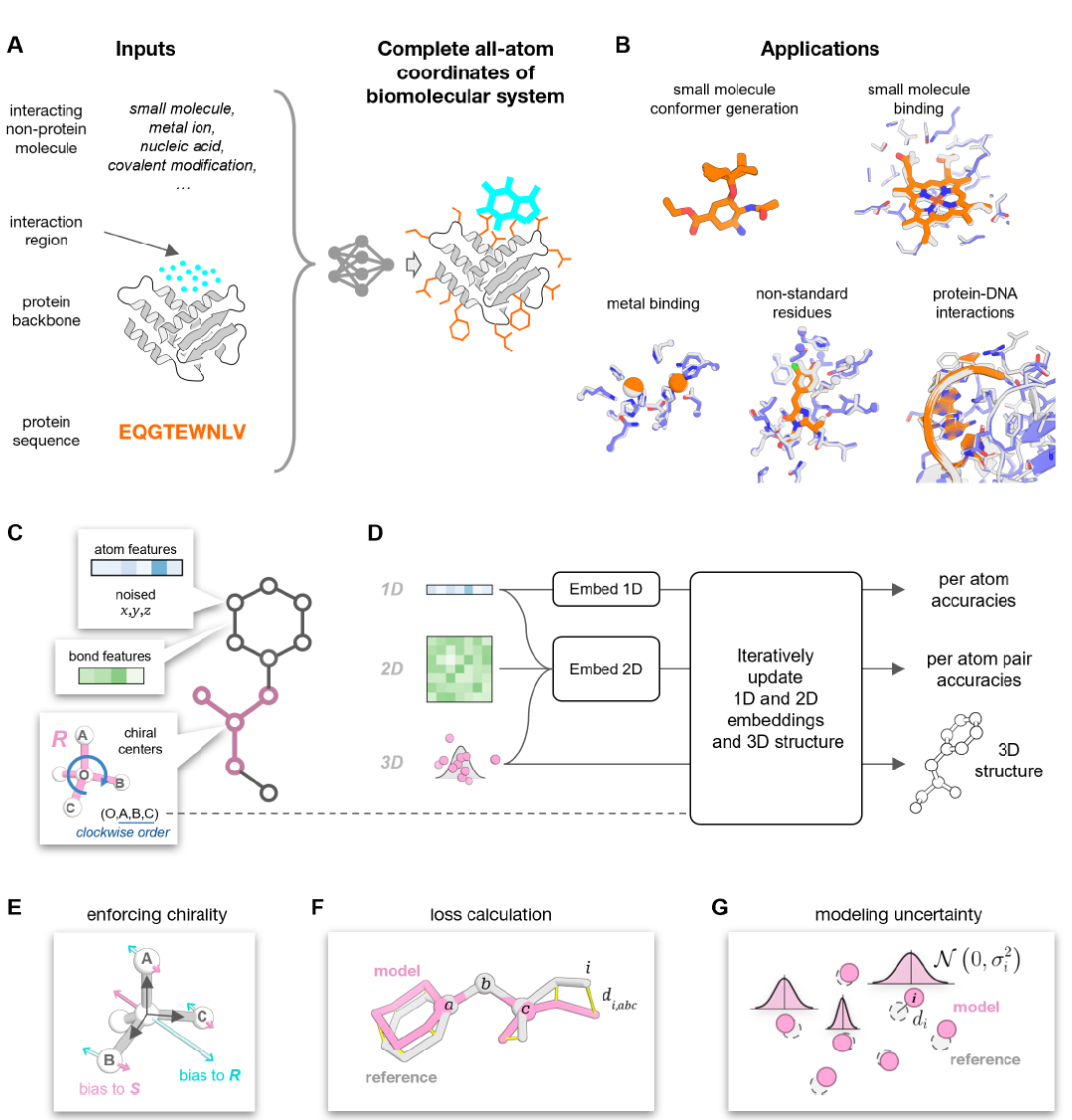
Lors de la phase d'entrée, le système moléculaire est transformé en un graphe chimique, où les nœuds représentent les atomes lourds individuels (les atomes d'hydrogène ne sont pas modélisés afin de réduire les coûts de calcul) et les arêtes représentent les liaisons chimiques entre les atomes (voir figure C ci-dessus). Cette représentation est cohérente pour différents types de molécules. Chaque nœud du réseau contient des informations sur le type d'atome et ses coordonnées 3D initialement perturbées. Le réseau a pour tâche de débruiter itérativement les coordonnées d'entrée tout en estimant simultanément les incertitudes sur les positions atomiques au sein de la structure du modèle de sortie (voir figure D ci-dessus).
PLACER adopte une architecture à trois pistes inspirée de RoseTTAFold (RF), et l'architecture réseau globale est la suivante :
* Conception à trois orbitales (1D, 2D, 3D) : les orbitales 1D gèrent les informations sur les caractéristiques atomiques ; les orbitales 2D gèrent les relations entre paires d'atomes (telles que les liaisons chimiques et la proximité spatiale) ; les orbitales 3D sont responsables de la mise à jour des coordonnées atomiques.
Optimisation itérative : Une fois l’intégration initiale des caractéristiques 1D et 2D terminée, ces dernières sont transmises au bloc d’itération pour mettre à jour itérativement les vecteurs d’intégration et la structure 3D. Dans ce bloc, un graphe de voisinage atomique est d’abord construit : pour chaque atome, la moitié de la proximité spatiale et l’autre moitié de la proximité chimique sont sélectionnées, soit un total de 32 atomes voisins les plus proches. Ensuite, les paires de caractéristiques 2D sont projetées en intégrations d’arêtes via une couche d’adaptation à propagation directe. Ces intégrations, combinées aux caractéristiques 1D, au graphe de voisinage atomique et à la structure atomique 3D actuelle, sont utilisées comme entrée du réseau SE3-Transformer pour mettre à jour les coordonnées 3D et les vecteurs d’intégration 1D.
Traitement du centre chiral : les informations relatives au centre chiral sont transmises au réseau via des caractéristiques de type 1 (vectorielles) (voir figure E ci-dessus). Les caractéristiques des orbitales 2D sont mises à jour par paires et combinées aux biais structuraux. Les fonctions de prédiction de confiance pour les atomes et leurs paires sont dérivées respectivement des orbitales 1D et 2D afin de finaliser le calcul des blocs itératifs. Le réseau entièrement entraîné comprend huit blocs itératifs à poids partagés.
Conception de la fonction de perte : L’entraînement de PLACER utilise une combinaison de perte structurelle et de perte de prédiction de confiance, appliquée après chaque itération. La perte structurelle principale est la FAPE (Frame-Aligned Point Error) calculée sur l’ensemble des atomes ; la confiance de la structure du modèle est évaluée aux niveaux atomique et par paire d’atomes.
Grâce à cette architecture réseau soigneusement conçuePLACER peut générer des ensembles diversifiés et atomiquement précis de conformations protéine-petite molécule à partir de coordonnées initialisées aléatoirement.Cela fournit une base fiable pour les analyses et la conception ultérieures. Contrairement aux méthodes de prédiction de la structure des protéines telles qu'AlphaFold,PLACER ne prédit pas la structure de la chaîne principale des protéines, mais se concentre sur les chaînes latérales au niveau atomique et les conformations des petites molécules, améliorant ainsi considérablement la vitesse de calcul et permettant la génération d'ensembles de conformations diversifiés.
Présentation des résultats : Fournir un soutien à l’ingénierie de précision, des petites molécules aux systèmes protéiques complexes
prédiction de la conformation des petites molécules
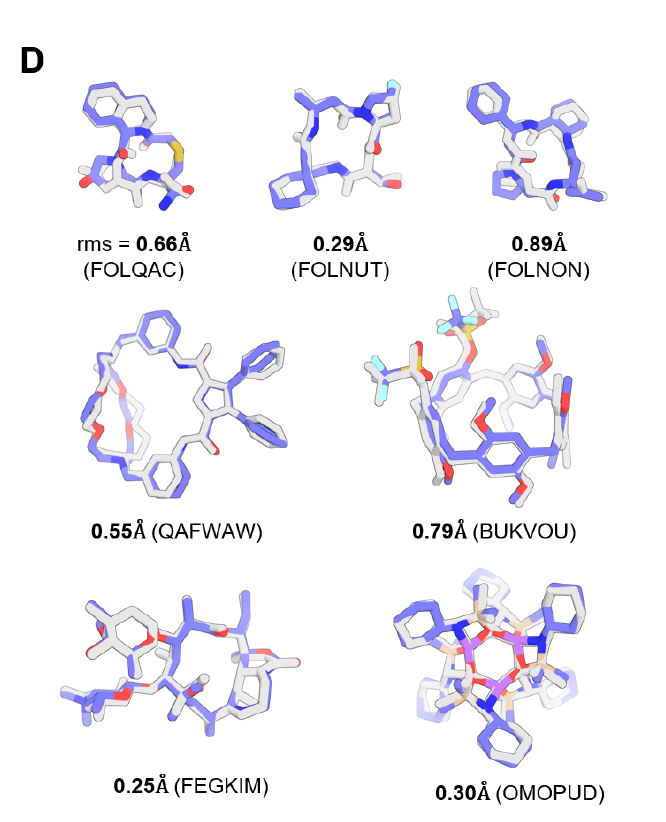
Les tests effectués sur l'ensemble de données de petites molécules du CSD démontrent que le PLACER entièrement entraîné peut générer correctement les structures tridimensionnelles de molécules complexes avec une précision inférieure à l'Å.On peut citer comme exemples les macrocycles comportant plus de 50 atomes (voir figure D ci-dessous), notamment les macrocycles peptidiques (voir la première rangée de la figure D ci-dessous). Les expériences d'ablation montrent que l'absence d'informations sur les distances de liaison ou la réduction du nombre d'itérations diminuent considérablement la précision des prédictions, ce qui souligne l'importance cruciale des stratégies d'itération et de caractéristiques conçues pour PLACER.
interactions protéine-petite molécule
Les chercheurs ont utilisé PLACER pour générer un ensemble de conformations de petites molécules dans la poche de la protéine cible (figure A ci-dessous) en exécutant le réseau plusieurs fois, chaque fois avec des initialisations aléatoires différentes des coordonnées d'entrée.L'analyse de l'ensemble de conformations généré montre que PLACER est insensible à la position initiale du ligand : plusieurs positions de départ différentes peuvent produire des prédictions proches de la conformation naturelle (figure B ci-dessous), et ces positions couvrent l'ensemble de l'espace de l'échantillonnage d'entrée (figure D ci-dessous).Les chercheurs ont également observé que le score RMSD prédit (pRMSD) calculé sur la base des atomes du ligand pouvait être utilisé pour sélectionner des modèles plus précis dans l'échantillon (figure E ci-dessous), le modèle ayant le score le plus élevé montrant un degré élevé de concordance avec la structure expérimentale (figure C ci-dessous).
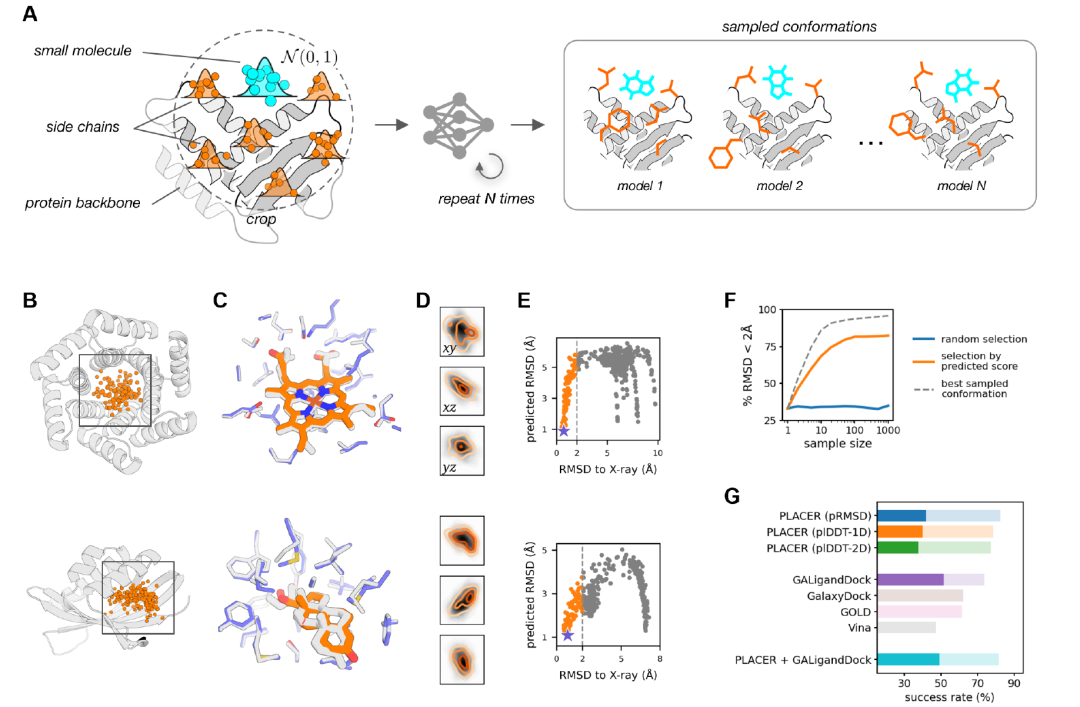
Comme illustré dans la figure G ci-dessus, lors du test de conformation non naturelle de 65 cibles médicamenteuses, PLACER a obtenu d'excellents résultats en générant et en sélectionnant des conformations quasi-natives, avec un taux de réussite, grâce au score pRMSD, supérieur à celui des outils d'amarrage traditionnels tels que Vina, GOLD et GalaxyDock. Comparé à la méthode Rosetta GALigandDock, la plus performante, PLACER a obtenu de meilleurs résultats dans la plage de faible précision (pourcentage de complexes avec un RMSD du ligand < 2 Å) (82,41 % contre 73,61 %), mais des résultats légèrement inférieurs dans la plage de haute précision (RMSD < 1 Å) (41,81 % contre 51,61 %).
Cependant, les performances de PLACER restent remarquables car, contrairement à d'autres méthodes, il n'est pas spécifiquement conçu pour les tâches d'amarrage protéine-petite molécule non naturelles. PLACER peut reconstruire la conformation des petites molécules et des chaînes latérales des protéines à partir de zéro, tandis que d'autres méthodes de test s'appuient principalement sur les coordonnées de la protéine d'entrée.
Conception du site actif enzymatique
L'application de PLACER à la conception de rétro-aldolases est particulièrement remarquable. L'équipe de recherche a réalisé 50 simulations répétées de la série RA95 de rétro-aldolases et de leurs versions améliorées, en analysant la diversité conformationnelle de la lysine du site actif et de son intermédiaire covalent. Les résultats ont montré que pour les enzymes présentant une faible activité lors de la conception informatique initiale, PLACER générait des ensembles conformationnels très diversifiés, indiquant une absence de pré-organisation ; tandis que pour les versions améliorées plus actives, les ensembles conformationnels devenaient de plus en plus ordonnés. Ceci suggère que…L’absence de pré-organisation constitue une lacune majeure dans la conception précoce des enzymes, tandis que PLACER fournit un outil d’évaluation rapide qui peut être utilisé pour orienter les travaux de conception des enzymes.
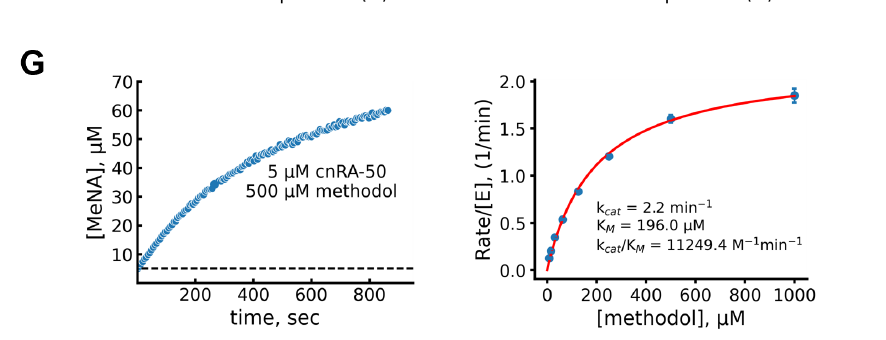
De plus, l'équipe a conçu un nouveau type d'enzyme d'inversion de l'aldostérone basé sur un repliement de type NTF2 et a évalué la corrélation entre le degré de préorganisation et la valeur kcat/KM à l'aide de PLACER. Les résultats ont montré que PLACER prédisait que les structures hautement préorganisées présentaient généralement une efficacité catalytique supérieure. La structure la plus active, cnRA-50, a atteint une valeur kcat/KM de 11 000 M⁻¹min⁻¹, nettement supérieure à celle des structures pré-organisées antérieures et proche de l'activité des structures créées à l'aide des méthodes RFdiffusion et proteinMPNN les plus récentes.
L'équipe de recherche prévoit que,Les méthodes de génération d'ensembles conformationnels basées sur PLACER seront largement utilisées pour la modélisation structurelle de molécules non protéiques complexes à l'état isolé ou dans des environnements protéiques, ainsi que pour l'évaluation de la conception d'enzymes et de conjugués protéine-petite molécule.
Professeur David Baker : Un pionnier qui s'est longtemps consacré à la conception informatique des protéines
Le 9 octobre 2024, le professeur David Baker, pionnier renommé de la conception de protéines, ainsi que Demis Hassabis et John M. Jumper de DeepMind, développeurs d'AlphaFold2, ont reçu le prix Nobel de chimie 2024.
Le professeur David Baker se consacre depuis longtemps à la conception computationnelle de protéines, en mettant à disposition des outils d'apprentissage profond en open source tels que RoseTTAFold, RFdiffusion et ProteinMPNN afin de faciliter la conception de nouvelles protéines. Il a également impulsé l'industrialisation de ces technologies en fondant une entreprise, ce qui fait de lui un véritable expert de renommée mondiale dans ce domaine. Dans ses dernières recherches, son équipe a réalisé des avancées majeures dans de nombreuses directions inédites.
Par exemple, lors du développement de nouveaux médicaments, les chercheurs utilisent souvent des protéines comme cibles principales, en liant le médicament à des protéines structurellement stables afin d'intervenir dans la progression de la maladie. Cependant, le ciblage des protéines désordonnées naturelles (IDP), qui ne possèdent ni structure, ni séquence, ni conformation préférentielle bien définies, demeure un défi. Dans ce contexte, en août 2025, l'équipe de David Baker a proposé Logos, une stratégie de conception de protéines basée sur une approche d'ajustement induit. Logos conçoit des protéines de liaison capables de s'adapter à 39 séquences d'acides aminés désordonnées cibles. Ainsi, un plus grand nombre de protéines peuvent servir de cibles pour le développement de nouveaux médicaments, ce qui pourrait accélérer la recherche sur le cancer et la maladie d'Alzheimer.
Titre de l'article :Conception de protéines de liaison aux régions intrinsèquement désordonnées
Adresse du document :https://www.science.org/doi/10.1126/science.adr8063
Le 18 septembre 2025, l'équipe de David Baker a proposé un modèle de diffusion atomique complet, RFdiffusion3 (RFD3), permettant la conception de novo d'interactions biomoléculaires atomiques. Ce modèle peut générer des structures protéiques en présence de ligands, d'acides nucléiques et d'autres groupements non protéiques. Plus simple et plus efficace que les méthodes précédentes, il a démontré, lors de plusieurs simulations informatiques comparatives, que RFdiffusion3 surpasse les méthodes antérieures, avec un coût de calcul dix fois inférieur.
Titre de l'article :Conception de novo d'interactions biomoléculaires tout-atome avec RFdiffusion3
Adresse du document :https://www.biorxiv.org/content/10.1101/2025.09.18.676967v1
Les canaux ioniques naturels jouent un rôle crucial dans les systèmes biologiques, et leurs versions artificielles sont largement utilisées dans les outils et capteurs chémogénétiques. Si la conception de protéines a permis de construire des protéines transmembranaires à structure poreuse, la conception de « filtres sélectifs » – c’est-à-dire des protéines dotées de chaînes latérales d’acides aminés précises ciblant des ions spécifiques – à l’instar des canaux ioniques naturels, constituait jusqu’à présent une limitation technologique. En octobre 2025, l’équipe de David Baker a, pour la première fois, utilisé l’intelligence artificielle pour concevoir un nouveau canal ionique calcique de novo. Cette étude démontre que même des fonctions biochimiques complexes, encore partiellement comprises, peuvent désormais être construites à partir de principes fondamentaux grâce à l’IA.
Titre de l'article :Conception ascendante des canaux Ca²⁺ à partir d'une géométrie de filtre de sélectivité définie
Adresse du document :https://www.nature.com/articles/s41586-025-09646-z
Au vu de leurs récents travaux, le professeur David Baker et son équipe redéfinissent à un rythme stupéfiant le paysage de la science des protéines : de la stratégie Logos, capable de se lier à des protéines naturellement désordonnées, à RFdiffusion3, qui permet la conception d’interactions moléculaires à l’échelle atomique, en passant par les recherches novatrices sur la première construction de novo de canaux ioniques calciques. L’équipe de Baker repousse sans cesse les limites de la conception computationnelle des protéines, de la théorie à la pratique. Leurs travaux élargissent non seulement les frontières de la conception biomoléculaire, mais rendent également l’avenir de la « construction de fonctions vitales par algorithmes » de plus en plus évident.
Liens de référence :
1.https://www.biorxiv.org/content/10.1101/2024.09.25.614868v2
2.https://www.thepaper.cn/newsDetail_forward_31663354
3.https://www.biorxiv.org/content/10.1101/2025.09.18.676967v1
4.https://www.nature.com/articles/s41586-025-09646-z








