Command Palette
Search for a command to run...
Prédiction De La Structure Des protéines/annotation Des fonctions/reconnaissance Des interactions/conception À La Demande, l'équipe Zhang Shugang De l'Université Océanique De Chine Touche Directement Aux Tâches Essentielles Du Calcul Intelligent Des Protéines
En tant que principaux vecteurs du vivant, les protéines jouent un rôle essentiel dans les fonctions physiologiques humaines. Cependant, la recherche traditionnelle est confrontée à des défis tels que le coût élevé de l'analyse structurale, un retard important dans l'annotation fonctionnelle et une faible efficacité dans la conception de nouvelles protéines. Ces dernières années, la demande d'analyse des propriétés complexes des protéines dans les sciences de la vie est devenue de plus en plus pressante. Le développement révolutionnaire de technologies telles que le big data, l'apprentissage profond et le calcul multimodal a ouvert de nouvelles perspectives pour la construction d'un système informatique intelligent pour les protéines. Ce système a permis aux protéines d'obtenir des résultats remarquables dans les domaines de l'annotation fonctionnelle à grande échelle, de la prédiction des interactions et de la modélisation structurale tridimensionnelle, ouvrant ainsi une nouvelle voie technique pour la découverte de médicaments et la simulation du vivant.
Lors de la conférence Zhiyuan de Pékin 2025, le professeur associé Zhang Shugang de l'École d'informatique de l'Université océanique de Chine a parlé du thème « Construction et application d'un système informatique intelligent à base de protéines » dans le forum « AI+Science & Engineering & Medicine ».En partant de la valeur fondamentale du système informatique intelligent des protéines, l'article explique systématiquement les avancées techniques de quatre tâches principales : la prédiction de la structure des protéines, l'annotation fonctionnelle, la reconnaissance des interactions et la nouvelle conception.Les résultats de recherche pertinents de l’équipe ont été mis en évidence.

HyperAI a compilé et résumé les propos approfondis du professeur associé Zhang Shugang sans en compromettre l'intention initiale. Voici la transcription de son discours.
Aperçu du système informatique intelligent des protéines : une révolution axée sur l'IA dans les sciences de la vie
Dans la recherche en sciences de la vie, l'importance des protéines est évidente. Non seulement elles sont une enzyme catalysant les réactions biochimiques, mais elles sont aussi un messager transmettant des signaux, constituant la base structurelle de l'organisme et l'« arme » du système immunitaire pour résister aux ennemis extérieurs. Cependant, les méthodes de recherche traditionnelles semblent impuissantes face aux caractéristiques complexes des protéines. Des problèmes tels que le coût élevé de l'analyse structurale, les retards importants dans l'annotation fonctionnelle et le faible taux de réussite de la conception des protéines sont devenus des défis majeurs.
L'introduction de l'IA a complètement inversé cette situation. En 2024, le prix Nobel de chimie a été décerné pour des avancées dans la prédiction et la conception de la structure des protéines par l'IA, démontrant ainsi une fois de plus pleinement le rôle crucial de l'IA dans la recherche sur les protéines.Le calcul intelligent des protéines permet une simulation et une prédiction efficaces des propriétés complexes des protéines en créant des modèles d'algorithmes basés sur les données.Il fournit également de nouvelles idées et de nouveaux paradigmes de recherche pour relever les défis ci-dessus et ouvre une nouvelle ère pour la recherche en sciences de la vie.
Percée dans la tâche principale de l'informatique intelligente des protéines
Les problèmes fondamentaux du calcul intelligent des protéines sont les quatre catégories suivantes :
La structure des protéines peut-elle être prédite à partir de zéro ?
Du paradoxe de Levinthal à la subversion d'AlphaFold
Prenons l’exemple du repliement des protéines : une protéine contenant 100 résidus peut avoir jusqu’à 10 conformations possibles.200 Si l'on effectue une recherche aléatoire, le temps nécessaire est bien supérieur à l'âge de l'univers (13,8 milliards d'années), ce qui constitue le célèbre paradoxe de Levinthal. Cependant, le repliement réel des protéines peut s'effectuer en quelques millisecondes ou minutes, ce qui suggère l'existence d'un chemin de repliement spécifique.
En 2018, la première génération du modèle AlphaFold a tenté de résoudre le problème à l'aide de méthodes d'apprentissage en profondeur, en utilisant le module de convolution résiduelle pour prédire la distance et l'angle de torsion des paires d'acides aminés.Dans CASP13, il a devancé ses concurrents avec une marge significative, prédisant avec précision 25 structures protéiques.Le deuxième n'a prédit que 3 bonnes réponses.
En 2021, le modèle de deuxième génération a réalisé un saut qualitatif. AlphaFold2 a utilisé HMMER et HH-suite pour effectuer l'alignement de séquences multiples et la recherche de modèles.Grâce à 48 modules Evoformer et 8 modules Structure, une prédiction de la structure des protéines avec une précision atomique est obtenue.Une base de données contenant environ 214 millions de prédictions de monomères protéiques a été publiée. L'erreur moyenne entre la structure prédite et les résultats de l'analyse par microscopie électronique ne dépasse pas la largeur d'un atome, ce qui correspond à la norme « hautement précise ».
En 2024, le modèle de troisième génération permettra de prédire pleinement la structure des interactions protéiques in vivo. AlphaFold3 a réalisé un bond qualitatif. Il peut non seulement prédire la structure des protéines,Il peut également prédire la structure de complexes composés de protéines, d’acides nucléiques, de petites molécules, d’ions et de toutes les autres molécules vivantes.Il couvre presque tous les types moléculaires de la base de données PDB, fournissant un outil puissant pour comprendre les fonctions cellulaires et le traitement des maladies.
Les fonctions des protéines peuvent-elles être annotées automatiquement : une avancée dans la fusion de données multi-sources
Grâce aux avancées d'AlphaFold3 dans le domaine de la prédiction des protéines, notre équipe a décidé d'orienter ses recherches vers l'annotation fonctionnelle des protéines et l'analyse des interactions. Actuellement, parmi les 250 millions de séquences protéiques mondiales, seule 0,51 TP3T a été annotée fonctionnellement avec précision. Le modèle traditionnel, basé sur l'analyse manuelle par des experts en biologie, s'est avéré incapable de gérer les données massives. Par conséquent, l'utilisation de l'apprentissage profond pour réaliser une annotation par lots à grande échelle est devenue une avancée majeure.
Notre exploration dans ce domaine a débuté en 2022. En visant le point sensible de l'industrie selon lequel les données structurelles de microscopie électronique, sur lesquelles repose l'apprentissage profond, sont rares et coûteuses,Nous proposons de manière innovante d'utiliser les données de structure virtuelle prédites par AlphaFold2 dans la formation des modèles.Cette stratégie, similaire à l'« enrichissement des données », a considérablement élargi l'échelle des données d'entraînement, passant des 5 millions d'échantillons fournis par les microscopes électroniques traditionnels à un bassin théorique de données de prédiction de plusieurs centaines de millions. La vérification expérimentale montre que le modèle entraîné à partir des données de prédiction non seulement surpasse la version native, mais permet également de découvrir de nouvelles fonctions protéiques non identifiées par les méthodes traditionnelles.
Titre de l'article :Amélioration des performances de prédiction de la fonction des protéines grâce aux structures protéiques prédites par AlphaFold
Adresse du document :
https://pubs.acs.org/doi/10.1021/acs.jcim.2c00885
En termes d’innovation technologique,Pour résoudre le problème de l'insuffisance d'exploration d'informations sur la structure des protéines, notre équipe a proposé une méthode de prédiction de la fonction des protéines basée sur l'attention graphique auto-supervisée.En codant les informations de corrélation des résidus dans la molécule de protéine et en utilisant pleinement les informations de distance entre les résidus comme tâche auxiliaire, les performances de la prédiction de la fonction protéique peuvent être améliorées.
Titre de l'article :SuperEdgeGO : Apprentissage de représentations graphiques supervisées par les bords pour une meilleure prédiction des fonctions protéiques (publication à venir)
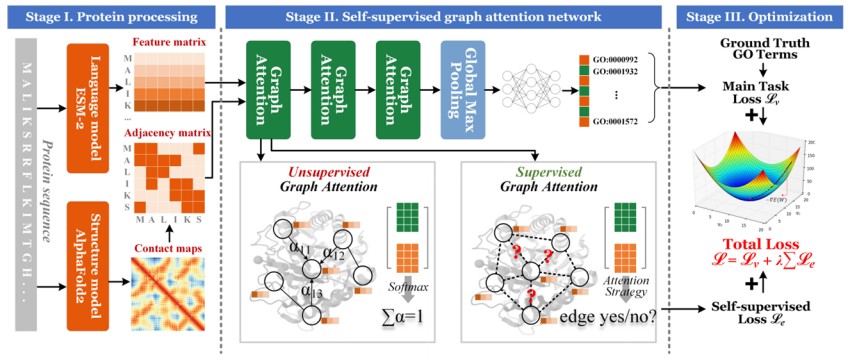
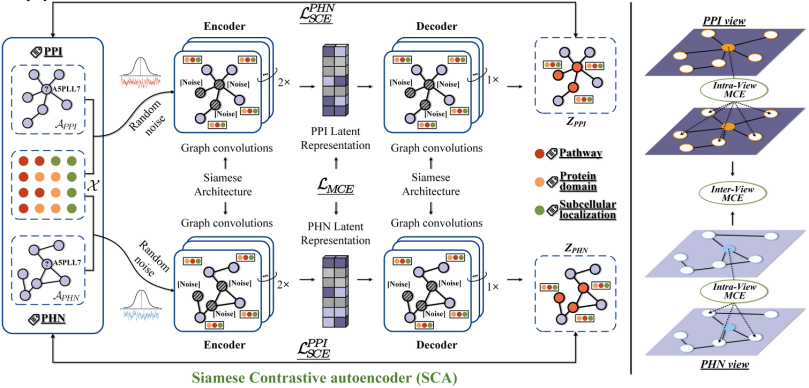
Pour résoudre les problèmes de caractéristiques protéiques hétérogènes difficiles à fusionner et spatialement incohérentes, une stratégie de construction de protéines à double vue et une méthode d'alignement des caractéristiques ont été proposées.Basé sur les caractéristiques complexes des protéines biologiques avec six modes transversaux (couvrant la séquence, la structure tridimensionnelle, le domaine fonctionnel et d'autres dimensions),L’équipe a également proposé une stratégie de fusion multimodale——Intégrer des méthodes d'apprentissage contrastif et d'analyse multi-vues dans le domaine informatique pour construire un modèle de fusion de caractéristiques hiérarchique. Cette solution a été comparée à 20 méthodes de référence courantes sur 7 ensembles de données, qui ont toutes obtenu des résultats SOTA, résolvant ainsi le problème technique de dégradation des performances causé par l'épissage direct des modalités.
Titre de l'article :Annotation des fonctions des protéines par la fusion de plusieurs modalités biologiques
Adresse du document :https://www.nature.com/articles/s42003-024-07411-y
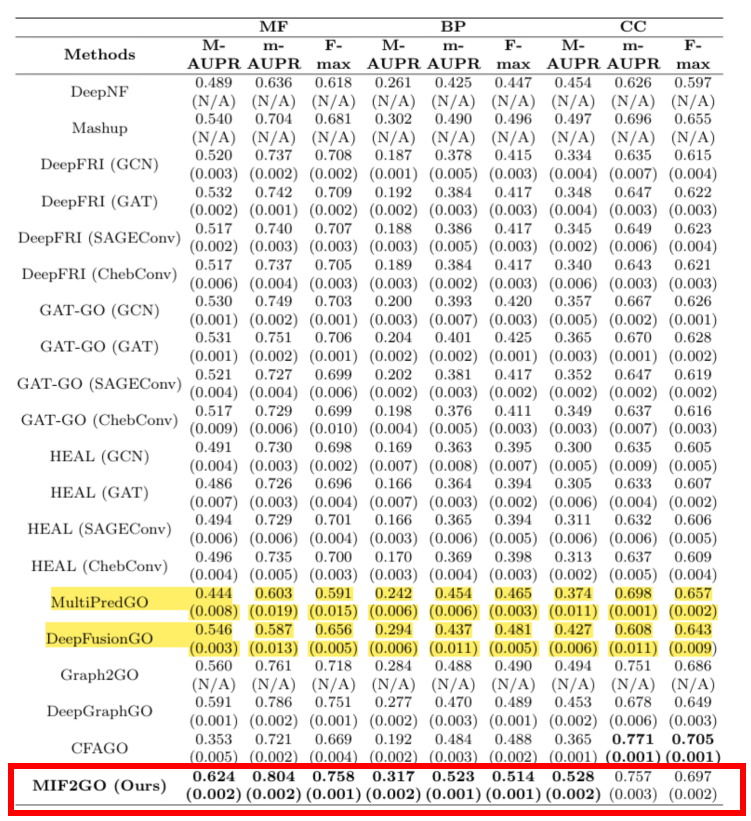
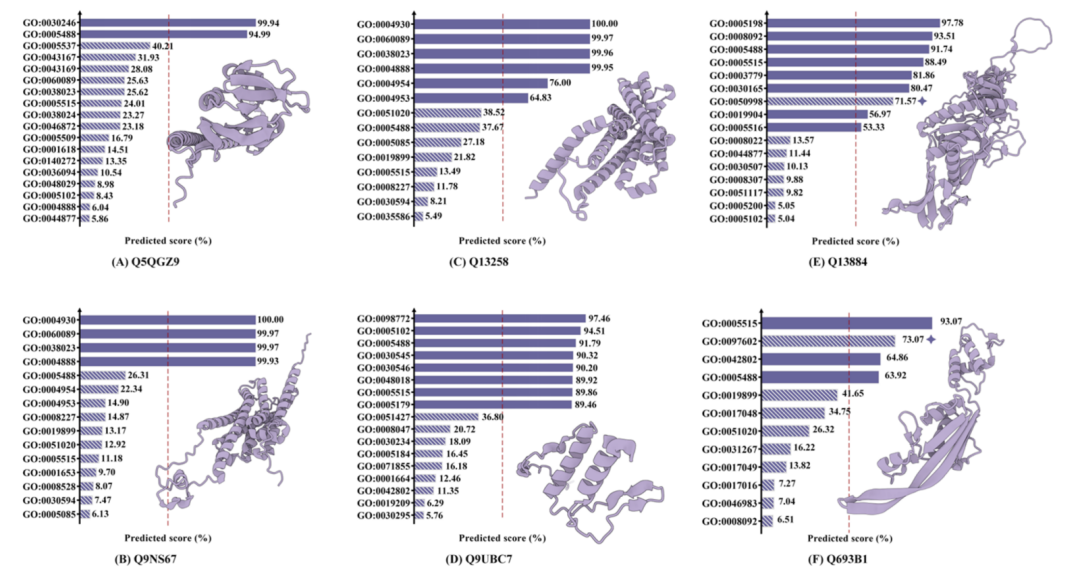
De plus, dans l’étude de l’interprétabilité de la prédiction fonctionnelle,Le modèle a également démontré une excellente capacité à identifier avec précision plus de 10 fonctions protéiques à partir de milliers d’annotations GoTerms.De plus, l'équipe a découvert, grâce à des recherches bibliographiques, que des cas où le modèle prédisait des erreurs tout en offrant un niveau de confiance élevé ont effectivement été recensés dans certaines études, ce qui indique que ces cas pourraient avoir été mal évalués en raison du décalage dans la version des données. Cette découverte souligne le potentiel des modèles d'IA pour explorer de nouvelles fonctions protéiques.
Les interactions protéiques peuvent-elles être identifiées avec précision ? Des modèles développés en interne permettent des prédictions efficaces.
Dans le domaine du développement de médicaments, l'arrimage précis de protéines comme cibles humaines est la clé de l'efficacité des médicaments, et l'IA a démontré une valeur importante dans ce processus. Bien qu'AlphaFold3 ait obtenu de bons résultats dans le domaine de la prédiction de la structure des protéines, ses applications pratiques présentent des limites évidentes : sa version gratuite ne prend en charge que 20 visites par jour, couvre environ 15 à 20 types de molécules, et il est extrêmement difficile d'obtenir des droits d'utilisation commerciale, ce qui a incité l'équipe à développer son propre modèle.
Sur la base de ce problème, l’équipe s’est concentrée sur les tâches suivantes :
Dans un premier temps, nous avons abordé les problèmes liés aux interactions synergétiques médiocres dans les méthodes existantes de prédiction des interactions protéiques.Un modèle d'apprentissage jumeau est introduit dans l'encodeur pour améliorer la cohérence collaborative de la représentation des protéines, et un cadre d'apprentissage collaboratif avec un mécanisme collaboratif d'interaction protéique et un mécanisme collaboratif de tâche est proposé.L’équipe a utilisé des méthodes d’attention interactive et d’apprentissage multitâche pour réaliser des prédictions interactives de protéines-acide nucléique, de protéines-protéines et de protéines-petites molécules.
L'équipe a également intégré des réseaux neuronaux Transformer et graph dans le domaine du PNL et a développé des modules tels que Convformer et Graphormer pour réaliser une modélisation interactive à distance.Le mécanisme d'attention croisée est utilisé pour renforcer la fusion des informations multimodales. Le modèle présente une forte capacité de généralisation dans des scénarios réels. Prenons l'exemple de la prédiction de la voie de signalisation du cancer du pancréas : son taux de précision dépasse 95%, avec seulement 9 paires d'erreurs de prédiction d'interaction.
Titre de l'article :SSPPI : Prédiction des interactions protéine-protéine améliorées par intermodalité à partir des perspectives de séquence et de structure (publication à venir)
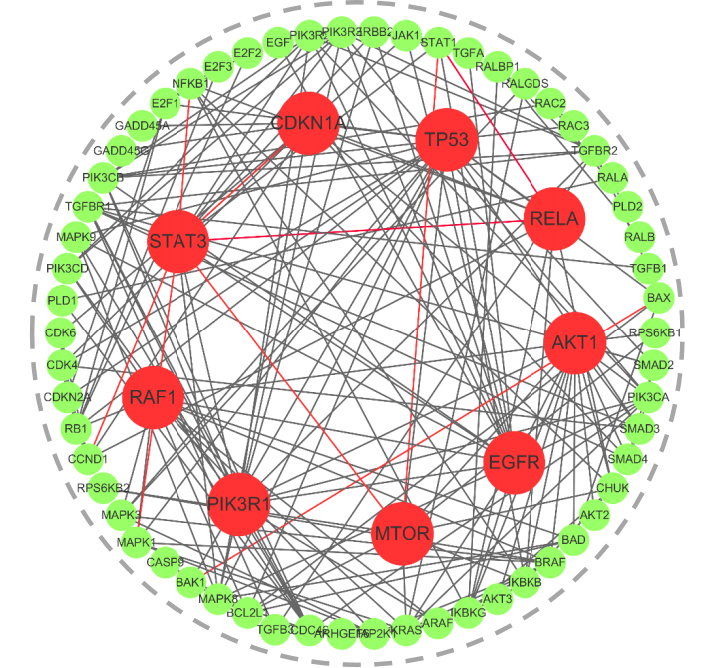
Ligne noire : prédiction correcte ; ligne rouge : prédiction erronée
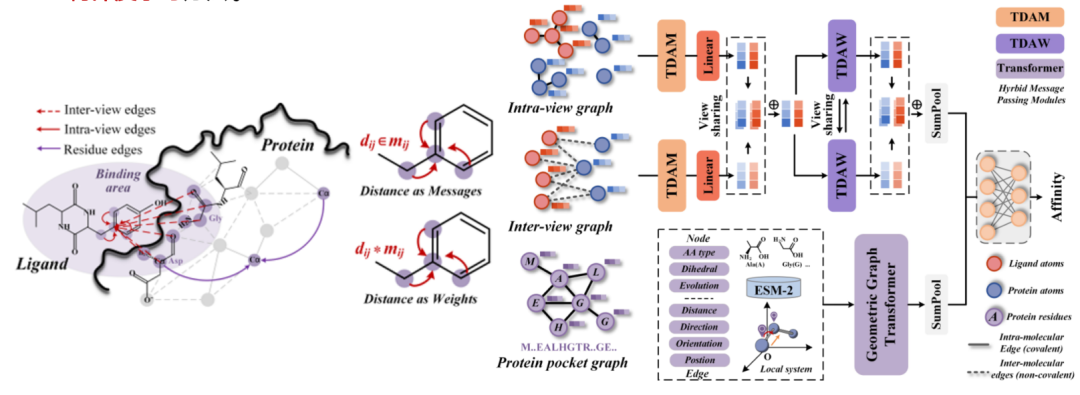
Dans des recherches récentes, outre la réduction dimensionnelle inter-échelle des protéines au niveau du réseau, nous nous sommes également attachés à exploiter les caractéristiques des protéines. Étant donné que les modèles de graphes traditionnels entraînent des pertes d'informations lors de la réduction d'informations structurelles tridimensionnelles à deux dimensions, nous avons introduit la dernière technologie d'apprentissage profond géométrique.Une méthode d'apprentissage profond géométrique basée sur une stratégie de transmission de messages hybride est proposée et un paradigme complet d'intégration d'informations tridimensionnelles est construit.Ce paradigme vise à résoudre l’irrationalité de l’élimination des informations tridimensionnelles dans la modélisation spatiale des sites et à fournir de nouvelles idées de recherche dans le domaine de la modélisation tridimensionnelle des protéines.
Titre de l'article :Apprentissage profond géométrique pour la prédiction d'affinité protéine-ligand avec des stratégies de transmission de messages hybrides (publication à venir)
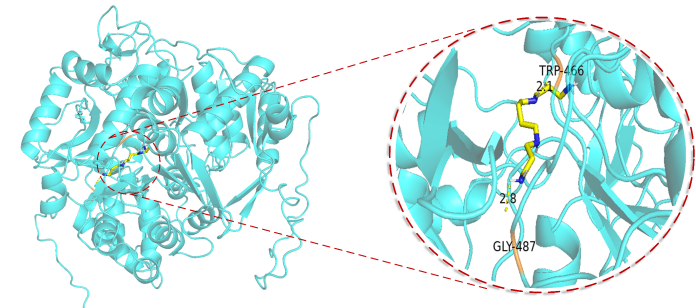
aussi,Nous avons également effectué des tests réels sur la protéine ACSS2 et sélectionné plusieurs composés candidats parmi des dizaines de milliers de composés.Les résultats de prédiction du modèle indiquent que l'affinité des composés criblés peut atteindre le niveau nM, montrant un bon potentiel médicamenteux ; notre équipe a coopéré avec l'équipe du Qingdao University Medical College pour effectuer la vérification, et les résultats d'amarrage ont également été confirmés de manière préliminaire dans les expériences humides menées récemment.
De nouvelles protéines peuvent-elles être conçues à la demande : des problèmes inverses aux applications innovantes
La conception des protéines est l'un des objectifs ultimes de la recherche sur les protéines et revêt une importance capitale pour le développement de vaccins, de traitements contre le cancer et de biomatériaux. Cependant, en tant que problème inverse du repliement des protéines, la conception des séquences protéiques est également confrontée à des défis tels que l'explosion de l'espace de recherche et les erreurs de simulation des champs de force traditionnels.
Face à la problématique fondamentale de la conception et de l'optimisation intelligentes des protéines, nous prenons ici comme exemple les derniers travaux de l'équipe de Baker, lauréate du prix Nobel l'an dernier. Le venin de serpent n'a pas d'antidote spécifique. Est-il possible de concevoir un nouveau type de protéine par ordinateur ? Face à ce problème, l'équipe de Baker a combiné ses précédentes expériences ProteinMPNN et RFDiffusion pour concevoir une nouvelle protéine. De plus, son équipe a également conçu des protéines de liaison spécifiques aux toxines du venin de serpent, offrant ainsi une nouvelle solution pour neutraliser les toxines mortelles du venin de serpent. L'article correspondant a été publié dans la revue Nature début 2025. Ces résultats de recherche démontrent le grand potentiel de l'IA dans le domaine de la conception des protéines et constituent une avancée majeure vers l'objectif de « création » de « conception de nouvelles protéines ».
Calcul multi-échelle des systèmes vivants complexes : simulation de la chaîne complète de l'échelle nanométrique à l'échelle macroscopique
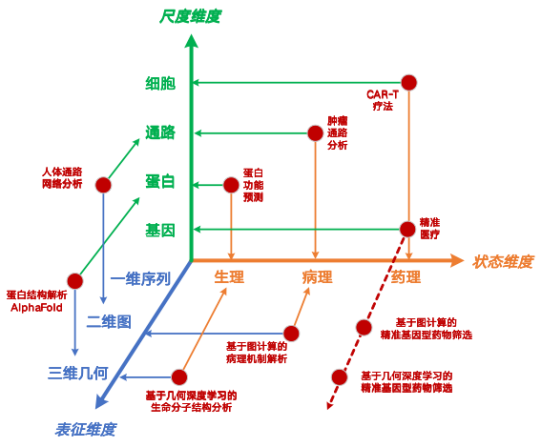
Le système vivant est un système complexe à plusieurs échelles. Du niveau nanoscopique des gènes au niveau macroscopique des cellules, chaque échelle interagit et s'influence mutuellement. Lors de ma visite au groupe de recherche du professeur Zhang Henggui à l'Université de Manchester, au Royaume-Uni, j'ai mené des recherches sur le cœur numérique. De retour en Chine, j'ai approfondi mes recherches sur les cellules numériques. Contrairement au paradigme de la « pulsion numérique » tel que le cœur numérique,L'équipe a proposé une méthode de modélisation multi-échelle pour les activités de la vie microscopique basée sur une approche de construction « pilotée par les données » et a construit un système de méthode de calcul microscopique tridimensionnel de « représentation-état-échelle ».Il couvre 36 points de recherche et il existe actuellement des articles ou des brevets accumulés sous près d'un tiers des méthodes.
De plus, sous la direction du professeur Wei Zhiqiang,Nous avons récemment défini le système de vie microscopique à quatre niveaux d’échelle.Incluant le niveau génétique nanoscopique, le niveau protéique « microscopique », le niveau de la voie de signalisation « mésoscopique » et le niveau cellulaire « macroscopique », la simulation du système de vie à chaîne complète est réalisée, dans l'espoir d'obtenir un couplage à grande échelle des atomes au cœur.
À propos du professeur associé Zhang Shugang
Zhang Shugang est professeur associé et directeur de master à l'École d'informatique de l'Université océanique de Chine, membre senior du CCF, membre correspondant du Comité de bioinformatique du CCF, membre du Comité de santé intelligente de la CAAI, directeur de la Shandong Bioinformatics Society et directeur de la National Natural Science Foundation of China, du Projet de dépenses d'entreprise de recherche scientifique fondamentale des universités centrales, etc. Il a été sélectionné pour le programme de soutien aux talents d'innovation postdoctorale du Shandong 2020.
Ses principaux domaines de recherche sont la biologie computationnelle et la bioinformatique, notamment la construction de cœurs numériques de très haute précision, la prédiction et la conception de la fonction des protéines, etc. Ces dernières années, il a publié plus de 30 articles dans des revues et conférences internationales faisant autorité telles que IEEE JBHI, JCIM et npj Systems Biology and Applications, avec plus de 1 600 citations sur Google Scholar.








