Command Palette
Search for a command to run...
Du Quartz Aux Matériaux Ferroélectriques, l'Université Harvard Propose Un Cadre d'apprentissage Automatique Équivariant Pour Accélérer La Simulation Du Champ Électrique À Grande Échelle Des Matériaux

En tant que domaine de recherche de pointe dans le domaine de la science des matériaux moderne, la science computationnelle des matériaux a pour mission essentielle d'analyser la microstructure des matériaux et de prédire leurs propriétés macroscopiques. S'appuyant sur les principes fondamentaux et les lois physiques fondamentales telles que la mécanique quantique, cette discipline s'attache à prédire avec précision les propriétés mesurables expérimentalement des matériaux réels, permettant ainsi une compréhension approfondie des mécanismes de réponse des matériaux aux stimuli externes. Ces caractéristiques de réponse comprennent les effets linéaires, non linéaires et de couplage.C'est l'élément central qui détermine les performances fonctionnelles des diélectriques, des ferroélectriques, des matériaux multiferroïques et des matériaux piézoélectriques.
Actuellement, la méthode de structure électronique des premiers principes basée sur la théorie de la fonctionnelle de la densité (DFT) est un moyen important pour étudier les propriétés des matériaux.Cependant, étant donné que le coût de calcul augmente de façon exponentielle avec la taille du système, il ne peut gérer que des systèmes à petite échelle.Cela a considérablement limité la recherche systématique sur les systèmes de matériaux complexes. Ces dernières années, l'introduction de méthodes d'apprentissage automatique a permis des avancées majeures dans ce domaine. Grâce à la construction de modèles basés sur les données, ces méthodes ont démontré un fort potentiel pour prédire la polarisation des matériaux, la charge de Born, la polarisabilité, la constante diélectrique, les caractéristiques spectrales, etc., et ont été appliquées avec succès aux molécules, à l'eau liquide et aux systèmes de matériaux solides.
Cependant, la plupart des méthodes d'apprentissage automatique existantes présentent encore des limites. Par exemple, il est difficile de garantir la stricte application des symétries physiques et des lois de conservation dans les modèles conçus indépendamment. Certaines solutions monomodèles, qui tentent d'intégrer les calculs de propriétés diélectriques, d'énergie et de force, rencontrent des difficultés lorsqu'elles sont étendues à des systèmes réels avec des conditions aux limites périodiques et des valeurs de polarisation multiples. Bien que certaines études aient construit des surfaces d'énergie potentielle en entraînant des forces atomiques sous différents champs électriques et en déduisant indirectement des caractéristiques de polarisation pour contourner la difficulté d'entraîner des données de polarisation multiples,Cependant, cette méthode repose sur des calculs de dérivées implicites, qui peuvent réduire la précision des prédictions, et le problème du coût élevé de l'acquisition de données DFT à grande échelle n'a pas été fondamentalement résolu.
Pour relever les défis ci-dessus, l'Université de Harvard et Robert Bosch LLC, une filiale du groupe allemand Bosch aux États-Unis, ont développé conjointement un cadre d'apprentissage différenciable unifié pour les réponses électriques.Ce cadre peut apprendre simultanément l’énergie potentielle généralisée et sa fonction de réponse aux stimuli externes dans un seul modèle d’apprentissage automatique.En définissant la fonction de réponse comme la dérivée de l'énergie potentielle généralisée par rapport aux coordonnées atomiques et aux paramètres de perturbation, et en utilisant la relation mathématique précise entre les deux, et en satisfaisant strictement les contraintes physiques telles que la conservation de l'impulsion et la règle de sommation acoustique de la charge de Born, les défauts inhérents aux modèles indépendants traditionnels sont surmontés, ouvrant une nouvelle voie pour la recherche de haute précision sur les propriétés diélectriques et ferroélectriques des matériaux cristallins, désordonnés et liquides.
Les résultats de recherche pertinents ont été publiés dans la revue de renommée internationale Nature Communications sous le titre « Apprentissage différentiable unifié de la réponse électrique ».
Points saillants de la recherche :
* Le premier cadre d'apprentissage automatique unifié permettant d'obtenir des garanties de conservation multidimensionnelles pour l'impulsion, l'enthalpie électrique, etc. sur la base de la relation différentielle précise entre l'énergie potentielle généralisée et les quantités de réponse observables.
* Développer un modèle de réseau neuronal équivariant pour briser le goulot d'étranglement de la simulation d'hystérésis ferroélectrique au niveau du million d'atomes et analyser avec précision la nucléation du domaine et la dynamique d'expansion unidimensionnelle de la commutation de polarisation.
* Résolvez le problème de la formation de polarisation à valeurs multiples et combinez les premiers principes pour obtenir une prédiction de haute précision à grande échelle des propriétés diélectriques et ferroélectriques dans le système α−SiO₂/BaTiO₃.

Adresse du document :
https://go.hyper.ai/18TWg
Autres articles sur les frontières de l'IA :
Expériences de données α−SiO₂ et BaTiO₃, construction d'un ensemble d'entraînement et dérivation des paramètres clés
Cette étude a mené des expériences de données sur deux matériaux : α-SiO₂ et BaTiO₃.Les performances du modèle sont vérifiées en construisant des ensembles d’apprentissage et des ensembles de validation.
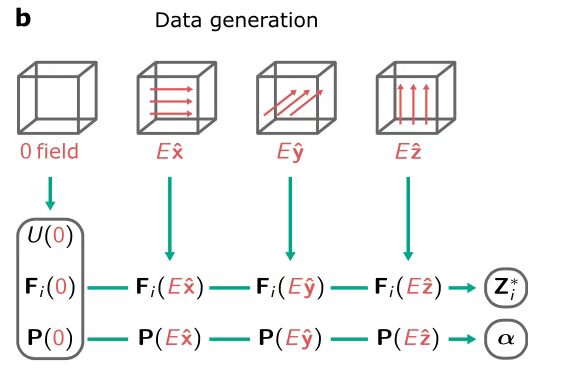
Français Dans la phase de génération de données d'entraînement, 200 images de configurations atomiques de α−SiO₂ ont été extraites de simulations MD classiques NVT à 300 K et 600 K (pilotées par le potentiel de Vashishta, chacune fonctionnant pendant 100 ps, échantillonnage par intervalle de 1 ps). Pour BaTiO₃, 75 images (300 K–400 K, 60 images de structure originale et 15 images de structure de paroi de domaine) ont été collectées grâce à la dynamique d'apprentissage actif du code FLARE.
Pour chaque ensemble de données, l'énergie, la force et la polarisation en l'absence de champ électrique ont été calculées par DFT, et la charge de Born et la polarisabilité ont été dérivées à l'aide de la méthode des différences finies sous un petit champ électrique de 0,36 MV/cm pour garantir la plage de réponse linéaire de la polarisation et du champ électrique.
Cadre de prédiction de la réponse matérielle basé sur un réseau neuronal équivariant
Dans cette étude, les chercheurs ont développé un cadre d'apprentissage automatique innovant visant à unifier l'apprentissage des fonctions d'énergie potentielle et de réponse généralisées. Ce cadre suit scrupuleusement les principes mathématiques du développement de Taylor.De manière ingénieuse, les tâches d’apprentissage de l’énergie potentielle généralisée et de ses dérivées sont réalisées simultanément dans un cadre de modèle unifié.Cela permet une prédiction précise des caractéristiques de réponse du matériau.
Le concept central de ce modèle est queEn prenant la dérivée de l'énergie potentielle généralisée par rapport à ses variables clés (telles que les positions atomiques et les champs externes), les caractéristiques de réponse correspondantes peuvent être générées automatiquement pour chaque configuration atomique.Il couvre de nombreux aspects tels que la force, la polarisation et la charge de Born. Cette conception garantit non seulement la mise en œuvre précise de la symétrie physique et des lois de conservation, telles que la conservation de la quantité de mouvement et de l'enthalpie électrique, mais améliore également considérablement la précision et la fiabilité des prévisions du modèle.
Dans la phase de formation du modèle,Les chercheurs y sont parvenus en ajoutant des paramètres décrivant les perturbations du système à l’entrée, permettant au modèle de différencier ces paramètres.De cette manière, l'entraînement peut être réalisé sur des quantités physiques supplémentaires. Parallèlement, les chercheurs ont minimisé une fonction de perte complète intégrant pleinement les facteurs de contribution de diverses caractéristiques de réponse telles que l'énergie, la force, la polarisation et la charge de Born. Ce mode d'entraînement est étroitement lié au concept d'entraînement de Sobolev. Plus précisément, chaque terme composant la fonction de perte est composé de la différence entre le gradient correspondant à l'étiquette d'entraînement et l'énergie.
Afin de capturer avec précision la dépendance non linéaire des caractéristiques de réponse sur plusieurs champs et les caractéristiques de couplage,Les chercheurs ont utilisé une architecture de réseau neuronal et une technique puissante appelée rétropropagation de gradient.Apprenez en profondeur le réseau complexe de relations entre les potentiels généralisés et leurs entrées.
Dans le processus de construction de l'architecture du modèle,Les chercheurs s’appuient sur la plateforme Allegro.Tirant pleinement parti de la haute précision et de l’efficacité exceptionnelle des données des réseaux neuronaux équivariants,Parallèlement, en suivant scrupuleusement le concept de conception locale, le modèle a atteint une excellente évolutivité. Concernant la conception des entrées du modèle, comme le montre la figure, les chercheurs ont intelligemment fusionné le champ électrique E avec la position atomique r_i, ce qui permet au modèle d'effectuer simultanément les tâches d'apprentissage de l'enthalpie électrique U, de la force F_i, de la polarisation P, de la charge de Born Z_i et de la polarisabilité α.
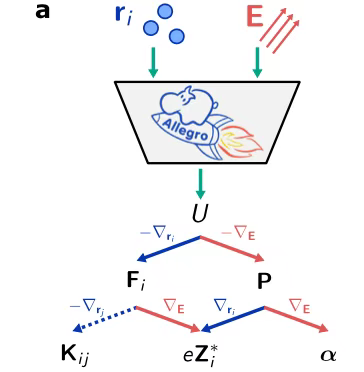
Dans le processus de génération de données de formation,Comme le montre la figure ci-dessous, les chercheurs ont utilisé des calculs DFT pour obtenir des données dans la zone autour du champ électrique nul et ont utilisé la méthode d'approximation des différences finies pour déterminer les valeurs de la charge de Born et de la polarisabilité. Après apprentissage, le modèle est capable de produire l'enthalpie, puis de dériver une série de paramètres clés tels que la force, la polarisation, la charge de Born et la polarisabilité en calculant les dérivées du premier et du second ordre de l'enthalpie de sortie.
Il convient de mentionner que les chercheurs ont spécialement développé une interface correspondante afin que le modèle puisse être intégré de manière transparente au logiciel LAMMPS, prenant ainsi fortement en charge les simulations de relaxation structurelle à grande échelle et de dynamique moléculaire d'apprentissage automatique (MLMD) dans des conditions de champ électrique.
Vérification multi-scénarios de la précision du modèle
Pour vérifier les performances du modèle, la recherche a mené des expériences dans deux directions principales : les vibrations des matériaux et les propriétés diélectriques, l’hystérésis ferroélectrique et la dynamique des dipôles.En prenant α−SiO₂ et BaTiO₃ comme objets de recherche, la précision et l'applicabilité du modèle dans différents scénarios sont explorées en profondeur.
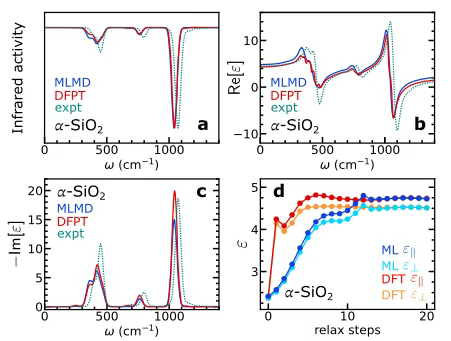
Dans l'étude des vibrations et des propriétés diélectriques,Les chercheurs ont choisi α−SiO₂ comme objet de recherche typique, ont extrait 200 images de données de 72 atomes issues de la simulation de dynamique moléculaire de l'énergie potentielle classique et ont entraîné le modèle d'apprentissage automatique correspondant sur cette base. Afin d'explorer plus avant la précision du modèle, une supercellule contenant 24 696 atomes a été construite. Après 10 picosecondes d'équilibre sous l'ensemble NVE, une simulation de dynamique moléculaire par apprentissage automatique (MLMD) de 200 picosecondes sans champ électrique a été réalisée. Le spectre infrarouge a été calculé par dynamique de polarisation, et la dynamique de polarisation et de polarisabilité a été analysée pour déterminer la constante diélectrique dépendante de la fréquence. Enfin, les résultats ont été comparés aux résultats de calcul basés sur la théorie des perturbations de la fonctionnelle de la densité (DFPT) et aux données expérimentales.
Comme le montre la figure ac ci-dessous,Les résultats du MLMD et du DFPT étaient très cohérents.De plus, lors de l'étude de l'effet de blindage des électrons et des ions en présence d'un champ électrique, les chercheurs ont effectué une relaxation structurale sous un champ électrique fini, basée sur la structure volumique d'origine de l'α-SiO₂. La constante diélectrique statique a été déterminée en calculant la différence de polarisation entre le champ électrique et l'absence de champ électrique. Les résultats sont présentés dans la figure d ci-dessous. Les constantes diélectriques statiques et à haute fréquence obtenues par le modèle sont globalement cohérentes avec les valeurs de la DFT.Cela démontre pleinement que le modèle peut capturer avec précision la contribution du champ électrique à la structure électronique, vérifiant ainsi son efficacité dans les simulations de dynamique de champ électrique fini.
Dans le même temps, lors de la vérification de la formation des charges de Born, il a été constaté que si le modèle n'est pas formé sur les charges de Born, la précision des vibrations et de la réponse diélectrique aux basses fréquences sera considérablement affectée, en particulier lorsque les données sont limitées, et le coût de calcul de la simulation sous champs électriques augmentera.Cette découverte souligne que la formation sur les charges Born est un avantage clé du modèle.
Dans l'étude de l'hystérésis ferroélectrique et de la dynamique dipolaire,Les chercheurs se sont concentrés sur la pérovskite BaTiO₃ et ont utilisé 75 images de données extraites par apprentissage dynamique actif, avec 135 atomes par image, pour entraîner le modèle d'apprentissage automatique. L'hystérésis ferroélectrique de la supercellule de 135 atomes a été calculée à température nulle, et les résultats sont présentés dans la figure a ci-dessous.L'hystérésis ferroélectrique obtenue par simulation MLMD est très cohérente avec celle obtenue par calcul DFT, ce qui vérifie fortement la fiabilité du modèle.
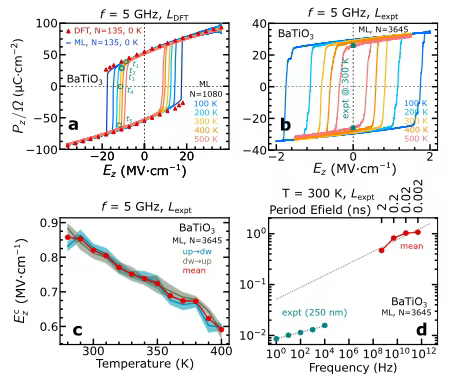
Afin d'évaluer plus en détail l'effet de la température sur la réponse ferroélectrique, les chercheurs ont réalisé une MLMD dans l'ensemble NVT sous un champ électrique sinusoïdal de 5 GHz. L'étude a révélé que, comme le montrent les figures bc ci-dessus,À mesure que la température augmente, le champ coercitif intrinsèque diminue, tandis que la polarisation spontanée est relativement moins affectée par la température.Que ce soit à température nulle ou à température finie, les courbes d'hystérésis présentent une symétrie de signe du champ électrique, cohérente avec les caractéristiques des champs vectoriels conservateurs de polarisation. De plus, lors de l'utilisation d'une supercellule de 3 645 atomes avec des paramètres de réseau expérimentaux pour la recherche, la polarisation spontanée est cohérente avec les résultats expérimentaux, et le champ coercitif est également plus proche de la valeur expérimentale, ce qui confirme la capacité du modèle à extrapoler sous des supercellules de numéros atomiques et de paramètres de réseau différents.
Lors de l'étude de l'effet de la fréquence du champ électrique sur l'hystérésis ferroélectrique,La MLMD d'une supercellule de 3 645 atomes à 300 K montre que le champ coercitif diminue avec la fréquence. Bien que la valeur extrapolée soit encore d'un ordre de grandeur différent de la valeur expérimentale, cette tâche de calcul complexe met en évidence les puissantes capacités de calcul du modèle et son excellente évolutivité vers des systèmes à millions d'atomes.
Recherche sur les modèles de matériaux : progrès collaboratifs entre le monde universitaire et l'industrie
Dans le domaine de la science des matériaux, les universités et le monde des affaires ont déployé de grands efforts pour promouvoir le développement continu de la recherche liée aux modèles de matériaux.
Pour la communauté universitaire, de nombreuses universités et équipes de recherche ont obtenu des résultats remarquables. Des chercheurs de l'Université de Rochester, aux États-Unis, ont développé un modèle d'apprentissage automatique capable d'analyser les données massives générées par les expériences de diffraction des rayons X (DRX) afin d'accélérer l'innovation dans le domaine des matériaux. Ce modèle est entraîné à partir de données expérimentales de matériaux inorganiques dans différentes conditions expérimentales et propriétés cristallines, puis classé et optimisé selon la loi de Bragg. Le modèle Chemeleon, proposé par l'Imperial College de Londres, utilise l'IA générative pour naviguer à partir de l'ensemble de données caractéristiques de la structure des matériaux, apprendre à partir de descriptions textuelles et de données de structure tridimensionnelle, et réaliser un échantillonnage de la composition chimique et de la structure cristalline. L'équipe collaborative de l'Université nationale de Séoul, en Corée du Sud, et de l'Université Fordham, aux États-Unis, a utilisé un modèle à grand langage (LLM) pour prédire la synthétisabilité de nouveaux matériaux et a expliqué les bases de cette prédiction, démontrant ainsi de bonnes performances et une bonne interprétabilité. L'équipe des professeurs Wang Jinlan et Ma Liang de l'École de physique de l'Université du Sud-Est en Chine et l'équipe du professeur Wang Xinran de l'Université de Nanjing ont proposé de nouvelles idées dans la recherche de matériaux bidimensionnels, réalisant la croissance épitaxiale contrôlée en nombre de couches minces MoS₂ à double couche uniformes de l'ordre du centimètre, et ont proposé des méthodes pour améliorer les performances des interfaces de contact grâce à la simulation informatique, obtenant avec succès une résistance de contact ultra-faible.
Le monde des affaires n'est pas en reste et s'engage activement dans l'innovation et l'application de la technologie des modèles de matériaux. Apple utilise des modèles d'apprentissage automatique pour optimiser les formules d'alliages métalliques destinés à la fabrication des coques de produits, améliorer la résistance et la durabilité des matériaux et réduire les coûts. BASF développe des modèles de matériaux avancés pour simuler leurs propriétés, accélérer le développement de nouveaux plastiques et revêtements et améliorer la compétitivité des produits. Le modèle MatterGen lancé par Microsoft génère des structures de matériaux répondant aux exigences de conception grâce à une architecture de modèle de diffusion unique, qui présente des avantages significatifs par rapport aux méthodes traditionnelles. L'entreprise a également collaboré avec l'équipe SIAT de l'Académie chinoise des sciences pour obtenir un nouveau matériau, le TaCr₂O₆.
Ces explorations de pointe et ces pratiques innovantes sont étroitement liées, faisant progresser sans cesse la recherche sur les modèles de matériaux. À l'avenir, grâce à l'approfondissement de la recherche et aux itérations et améliorations technologiques, les modèles de matériaux connaîtront des avancées et des applications dans un plus large éventail de domaines, jetant ainsi des bases solides pour le développement scientifique et technologique.
Articles de référence :
1.https://mp.weixin.qq.com/s/mctu0DOWO_OieLnOgp93Rw
2.https://mp.weixin.qq.com/s/I-UZTyUFSWwXlf1LCmwjRQ
3.https://mp.weixin.qq.com/s/Ox62ut3IJcUWsLC7sF100Q
4.https://mp.weixin.qq.com/s/VlPb8zSghVVxnPNl-WzqBA
5.https://plastics-rubber.basf.com/global/en/performance_polymers/services/service_ultrasim/Material-Modeling








