Command Palette
Search for a command to run...
Entraîné Avec Moins De 100 000 Points De Données Structurées, l'École Polytechnique Fédérale De Lausanne (EPFL) a Proposé PET-MAD, Atteignant Une Précision De Simulation Atomique Comparable Aux Modèles professionnels.
Des matériaux semi-conducteurs aux molécules médicamenteuses actives, la structure électronique demeure essentielle à la compréhension de leurs performances. Les calculs ab initio, basés sur la mécanique quantique, permettent de prédire avec précision la structure, la stabilité et la fonction de la matière, et ont été à l'origine de progrès rapides dans la conception des matériaux et le développement des médicaments. Cependant, la complexité des calculs augmente considérablement avec la taille du système.Même les supercalculateurs les plus performants peinent à simuler des processus complexes tels que le repliement des protéines et les réactions catalytiques sur de longues périodes.Cela a créé une situation où « le mécanisme peut être décrit, mais il est difficile à calculer ».
Alors que les méthodes traditionnelles atteignent leurs limites de calcul, l'apprentissage automatique offre une nouvelle voie pour les simulations à l'échelle atomique. L'apprentissage automatique des potentiels interatomiques, entraîné sur des données de mécanique quantique, permet de modéliser des structures électroniques complexes avec efficacité, réduisant ainsi les coûts de calcul de plusieurs ordres de grandeur tout en conservant une précision proche de celle des calculs ab initio. Au cours des deux dernières décennies, il a démontré des avantages significatifs dans des systèmes tels que les alliages à haute température et les biomacromolécules, en palliant les limitations liées à l'imprécision des champs de force empiriques et au coût prohibitif des méthodes ab initio. Cependant,Les premiers modèles étaient principalement conçus pour des systèmes uniques, ce qui nécessitait la génération répétée de données d'entraînement et le réajustement des paramètres, rendant la recherche intersystème assez difficile.
Par conséquent, la généralisation est devenue un axe de développement majeur. Ces dernières années, divers modèles généraux adaptables à différents éléments et environnements chimiques ont vu le jour.Cependant, en raison de données d'entraînement insuffisantes et de normes d'évaluation incohérentes, ses performances globales restent instables.
Dans ce contexte, le modèle PET-MAD proposé par l’École polytechnique fédérale de Lausanne (EPFL) fournit de nouveaux échantillons.En exploitant un ensemble de données couvrant un large éventail de diversité atomique et une architecture de réseau basée sur le Point Edge Transformer, il atteint une précision comparable aux modèles dédiés PET-Bespoke tout en utilisant beaucoup moins d'échantillons d'entraînement que les modèles traditionnels.Ceci constitue un exemple éloquent du développement de la simulation atomique vers une plus grande efficacité et une applicabilité plus large.
Les résultats de recherche pertinents, intitulés « PET-MAD comme potentiel interatomique universel léger pour la modélisation avancée des matériaux », ont été publiés dans Nature Communications.

Adresse du document :
https://www.nature.com/articles/s41467-025-65662-7
Suivez notre compte WeChat officiel et répondez « PET-MAD » en arrière-plan pour obtenir le PDF complet.
Autres articles sur les frontières de l'IA :
https://hyper.ai/papers
Ensemble de données MAD : contient 85 éléments et près de 100 000 structures
L'ensemble de données MAD a été construit pour fournir une base de données de haute qualité pour l'entraînement des potentiels interatomiques (MLIP) dans l'apprentissage automatique général, qui présente une grande diversité chimique, une cohérence stricte des données et une bonne facilité d'utilisation.Il contient 85 éléments dont les numéros atomiques vont de 1 à 86 (à l'exclusion de l'astate), soit un total de 95 595 structures.Il est systématiquement divisé en 8 sous-ensembles en fonction de ses caractéristiques chimiques et structurelles, couvrant globalement les cristaux massifs, les surfaces, les agrégats, les matériaux bidimensionnels et les systèmes moléculaires.
Le cœur de l'ensemble de données provient d'un sous-ensemble de la base de données Materials Cloud 3D, MC3D.Elle contient 33 596 structures cristallines de phases individuelles. À partir de celles-ci, un sous-ensemble MC3D perturbé (30 044 structures) a été généré en perturbant les coordonnées atomiques par un bruit gaussien, et un sous-ensemble MC3D aléatoire (2 800 structures) a été construit en réarrangeant aléatoirement les types d'atomes dans le cristal. Ces deux sous-ensembles sont spécifiquement conçus pour améliorer la couverture des configurations de distorsion extrêmes ou hors équilibre.
Pour répondre aux exigences de simulation des surfaces et des systèmes de basse dimension, l'ensemble de données comprend le sous-ensemble MC3D-surface (5 589 modèles de plans cristallins à faible indice) et le sous-ensemble MC3D-cluster (9 071 nanoclusters) obtenus par découpage de phase massive, ainsi que le sous-ensemble MC2D (2 676 cristaux bidimensionnels) importé de la base de données Materials Cloud 2D. Concernant les systèmes moléculaires, il est complété par deux sous-ensembles : SHIFTML-molcrys (8 578 cristaux moléculaires) et SHIFTML-molfrags (3 241 fragments moléculaires), tous issus de bases de données de référence afin de garantir la fiabilité des échantillons.
Concernant les stratégies de génération de données,MAD combine la réutilisation directe et la modification structurale. Quatre sous-ensembles — MC3D, MC2D, SHIFTML-molcrys et SHIFTML-molfrags — utilisent directement des structures publiées, mais recalculent les énergies et les forces à l'aide de paramètres DFT identiques, éliminant ainsi les biais systématiques entre les différentes sources. Les autres sous-ensembles, par des modifications physiques de la structure de base (ajout de bruit, réarrangement d'atomes, découpe de surfaces, etc.), étendent de manière ciblée la diversité des configurations.
PET-MAD : Architecture et performances remarquables d’un modèle de potentiel interatomique universel
Le modèle PET-MAD est entraîné sur une architecture optimisée par la frontière de Pareto.L'architecture se compose de deux couches de passage de messages, chacune équipée de deux sous-couches Transformer, utilisant des représentations de jetons de 256 dimensions, un mécanisme d'attention multi-têtes à 8 têtes, et produisant finalement le résultat via une couche entièrement connectée avec 512 neurones.Pour améliorer la stabilité du processus d'entraînement, avant l'entraînement formel, le système ajustera un modèle linéaire et soustraira la contribution de base de l'énergie liée à la composition atomique.
L'entraînement complet a été réalisé avec le framework PyTorch et le package Metatrain. Les calculs ont été répartis sur huit GPU NVIDIA H100, avec une taille de lot de 24 architectures par GPU. L'entraînement a duré environ 40 heures, soit 1 500 époques. L'optimisation a utilisé l'optimiseur Adam, avec un taux d'apprentissage initial de 10⁻⁴, divisé par deux toutes les 250 époques. La fonction de perte correspond à l'erreur quadratique moyenne des prédictions d'énergie et de force, le terme d'énergie étant pondéré à 0,1.
Après l'entraînement, le modèle a atteint une erreur absolue moyenne de 7,3 meV/atome pour l'énergie et de 43,2 meV/Å pour la force sur l'ensemble d'entraînement, et de 14,7 meV/atome pour l'énergie et de 172,2 meV/Å pour la force sur l'ensemble de validation, démontrant une excellente précision d'ajustement.
Pour permettre au modèle général de mieux s'adapter à des systèmes chimiques spécifiques,PET-MAD introduit la technologie de réglage fin d'adaptation de bas rang (LoRA).Contrairement au réglage fin traditionnel de tous les paramètres, qui peut facilement entraîner un « oubli catastrophique » (c’est-à-dire une perte des connaissances générales existantes), LoRA fige tous les poids du modèle de base, n’injectant que deux paires de matrices de faible rang entraînables dans chaque module d’attention et ajustant leur influence via un paramètre d’échelle, atteignant ainsi l’objectif d’une « amélioration précise sans perte de généralité ». Dans cette étude, le rang de LoRA est fixé à 8 et le paramètre d’échelle est fixé à 0,5.
Les expériences montrent queDans les scénarios où les données sont limitées, les modèles affinés avec LoRA surpassent systématiquement les modèles dédiés entraînés à partir de zéro pour ce système.Même dans des systèmes complexes tels que le titanate de baryum, des modèles finement réglés peuvent produire des observables physiques comparables à celles de modèles dédiés tout en conservant une précision générale, et sont donc recommandés comme schéma d'optimisation pour des applications spécifiques.
Pour les modèles à usage général, une estimation fiable de l'erreur est cruciale. PET-MAD quantifie l'incertitude grâce à la méthode LLPR (Last Layer Prediction Rigidity). Cette méthode estime l'erreur a posteriori des nouvelles prédictions en analysant la covariance de la dernière couche de caractéristiques cachées du modèle sur l'ensemble d'entraînement, sans coût de calcul supplémentaire significatif. Plus important encore, LLPR permet d'échantillonner un nombre fini de poids de la dernière couche pour former un « ensemble superficiel » léger, transférant ainsi l'incertitude au calcul de quantités dérivées complexes telles que l'énergie libre et les courbes de dispersion des phonons. Cette méthode présente un coût de calcul nettement inférieur aux méthodes d'ensemble traditionnelles, ce qui la rend parfaitement adaptée aux besoins pratiques des simulations atomiques à grande échelle.
aussi,PET-MAD propose deux modes de prédiction de la force : l’un est basé sur la différenciation automatique (rétropropagation) pour déduire la force à partir de l’analyse énergétique, ce qui satisfait strictement la conservation de l’énergie ; l’autre consiste à prédire la force directement à partir des coordonnées atomiques grâce à un module de réseau neuronal séparé, ce qui peut améliorer la vitesse d’inférence de 2 à 3 fois, mais peut introduire une petite quantité de non-conservation de l’énergie.En concevant un intégrateur dédié à plusieurs pas de temps, l'équipe de recherche a efficacement évité les erreurs d'échantillonnage qui pourraient être causées par ce dernier mode, assurant ainsi à la fois l'efficacité de calcul et la fiabilité de la simulation dynamique.
Avantages à la fois théoriques et pratiques : obtenir la précision d’un modèle dédié avec un minimum de données propriétaires.
Afin d'évaluer de manière exhaustive les performances de PET-MAD, l'équipe de recherche l'a d'abord comparé à des systèmes modèles de référence tels que MACE-MP-0 et Orb-v2 à l'aide de tests de performance faisant autorité, comme Matbench Discovery. Pour garantir l'équité des résultats, les chercheurs ont recalculé un sous-ensemble de tests de référence en utilisant des paramètres DFT compatibles avec PET-MAD afin d'éliminer les biais liés aux différences de méthodes de calcul.
Les résultats des tests sont présentés dans le tableau ci-dessous.PET-MAD, entraîné avec moins de 100 000 structures (1 à 3 ordres de grandeur de données en moins que les modèles similaires), et avec un nombre modéré de 2,8 millions de paramètres (bien inférieur aux 25 millions d'Orb-v2 et aux 15,8 millions de MACE-MP-0-L), surpasse tous les modèles comparables sur les ensembles de données moléculaires (SPICE, MD22).Il est comparable à des modèles de pointe tels que MatterSim-5M dans les ensembles de données inorganiques, et ses avantages sont plus significatifs sur les ensembles de données avec des configurations fortement déformées, certains modèles ayant une erreur 50 fois supérieure à celle de leurs homologues.
Cependant, les tests de référence ne suffisent pas à vérifier pleinement la fiabilité du modèle dans des scénarios complexes. C’est pourquoi l’équipe de recherche a sélectionné six cas d’application divers et exigeants afin de comparer les performances de PET-MAD, de son modèle d’ajustement LoRA et du modèle dédié PET-Bespoke.
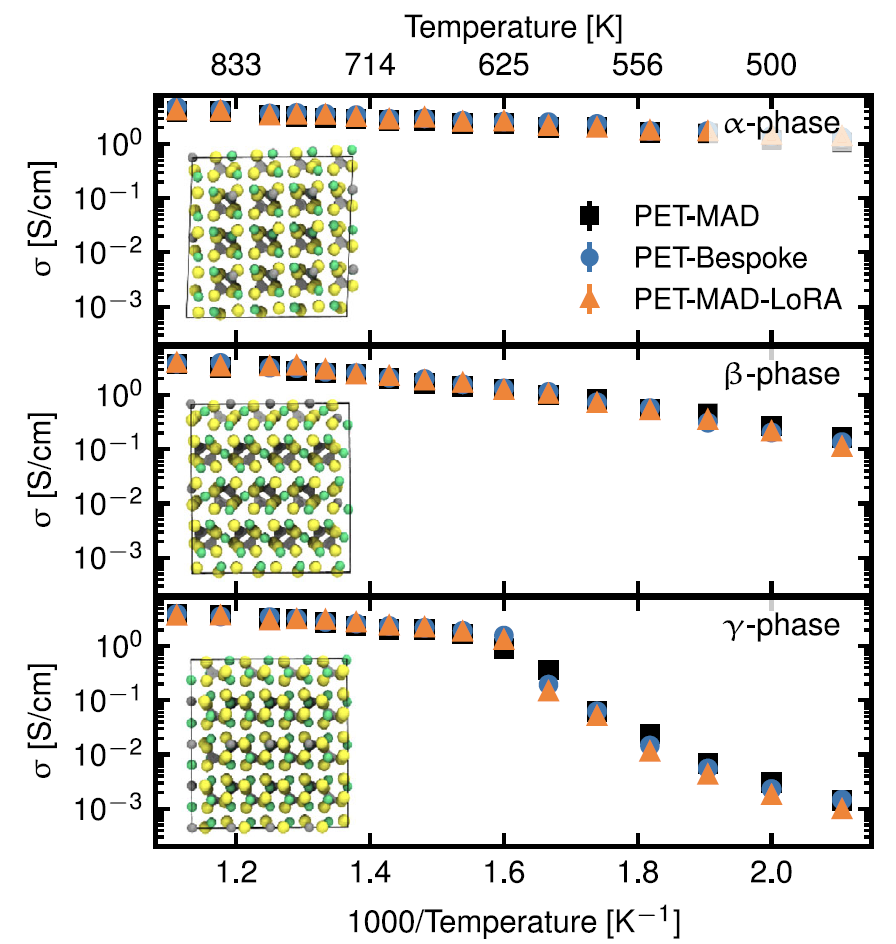
1. Électrolyte solide : thiophosphate de lithium (LPS)
L'étude porte sur sa principale caractéristique de performance : la conductivité ionique. Comme le montre la figure ci-dessous, bien que les erreurs de vérification du PET-MAD en termes d'énergie et de force (4,9 meV/atome, 163,9 meV/Å) soient supérieures à celles du modèle dédié PET-Bespoke (1,2 meV/atome, 35,6 meV/Å).Cependant, la conductivité prédite par les simulations de dynamique moléculaire qui la pilotent est en parfait accord avec les résultats du modèle dédié PET-Bespoke et du modèle finement ajusté sur une large plage de températures.Il a capturé avec précision l'effet des motifs structuraux (rotation tétraédrique PS₄) sur le transport des ions, avec seulement une légère surestimation de la température de transition de phase de la phase γ.
2. Matériau semi-conducteur : arséniure de gallium (GaAs)
Les résultats du calcul du point de fusion par la méthode d'ancrage d'interface sont présentés dans la figure ci-dessous. Le PET-MAD ajusté par LoRA concorde parfaitement avec les résultats de prédiction du modèle dédié PET-Bespoke (1169 ± 3 K contre 1169 ± 4 K).
Bien que la valeur prédite du modèle général pré-entraîné (1111±72 K) soit légèrement inférieure, son erreur est beaucoup plus petite que l'écart inhérent de la fonctionnelle DFT elle-même par rapport à la valeur expérimentale (1511 K).Ce cas met également en évidence l'intérêt de la fonction intégrée de quantification de l'incertitude LLPR du PET-MAD, qui permet d'évaluer et de transmettre les erreurs à faible coût, fournissant ainsi une référence pour la fiabilité des résultats.
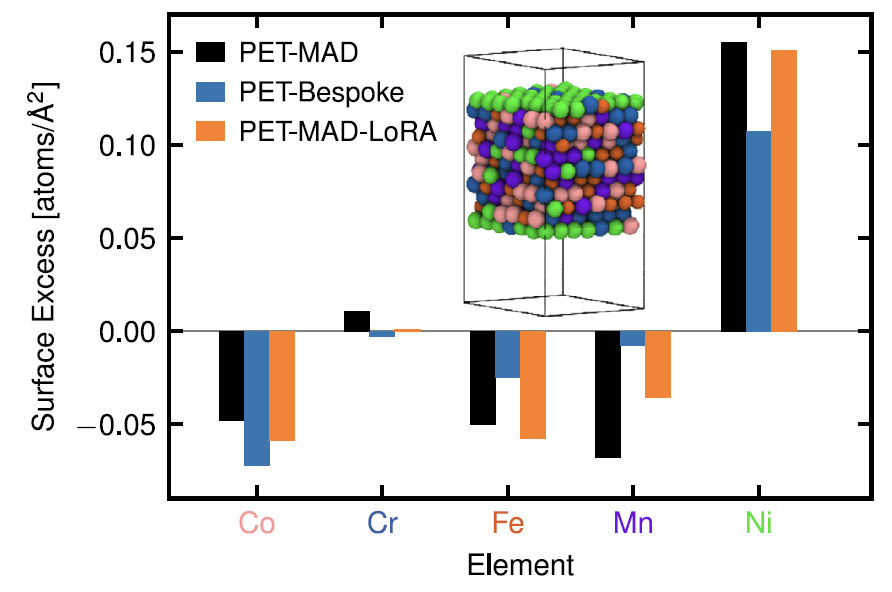
3. Alliages à haute entropie : CoCrFeMnNi
En ce qui concerne son comportement de ségrégation élémentaire en surface, l'étude a constaté, comme le montre la figure ci-dessous, que le PET-MAD pré-entraîné peut reproduire avec précision les caractéristiques de ségrégation d'une « surface riche en nickel et appauvrie en autres éléments » sans formation spéciale pour ce système, ce qui est en accord quantitatif avec l'étude de référence.
Bien que son erreur énergétique (25,8 meV/atome) soit plus élevée que celle du modèle dédié PET-Bespoke (14,6 meV/atome).Cependant, ce dernier peut être exposé à un risque de surapprentissage en raison de données d'entraînement limitées (seulement 2000 structures), tandis que PET-MAD montre une meilleure robustesse.
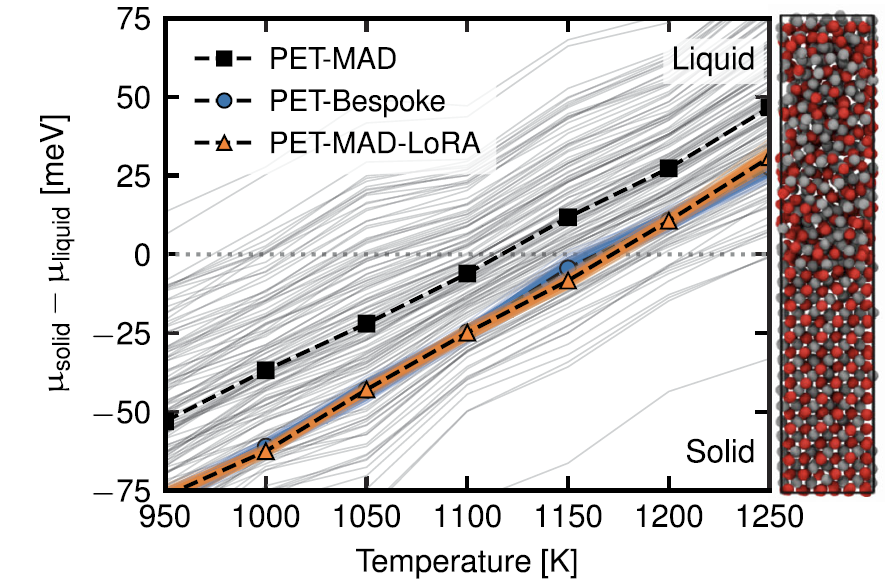
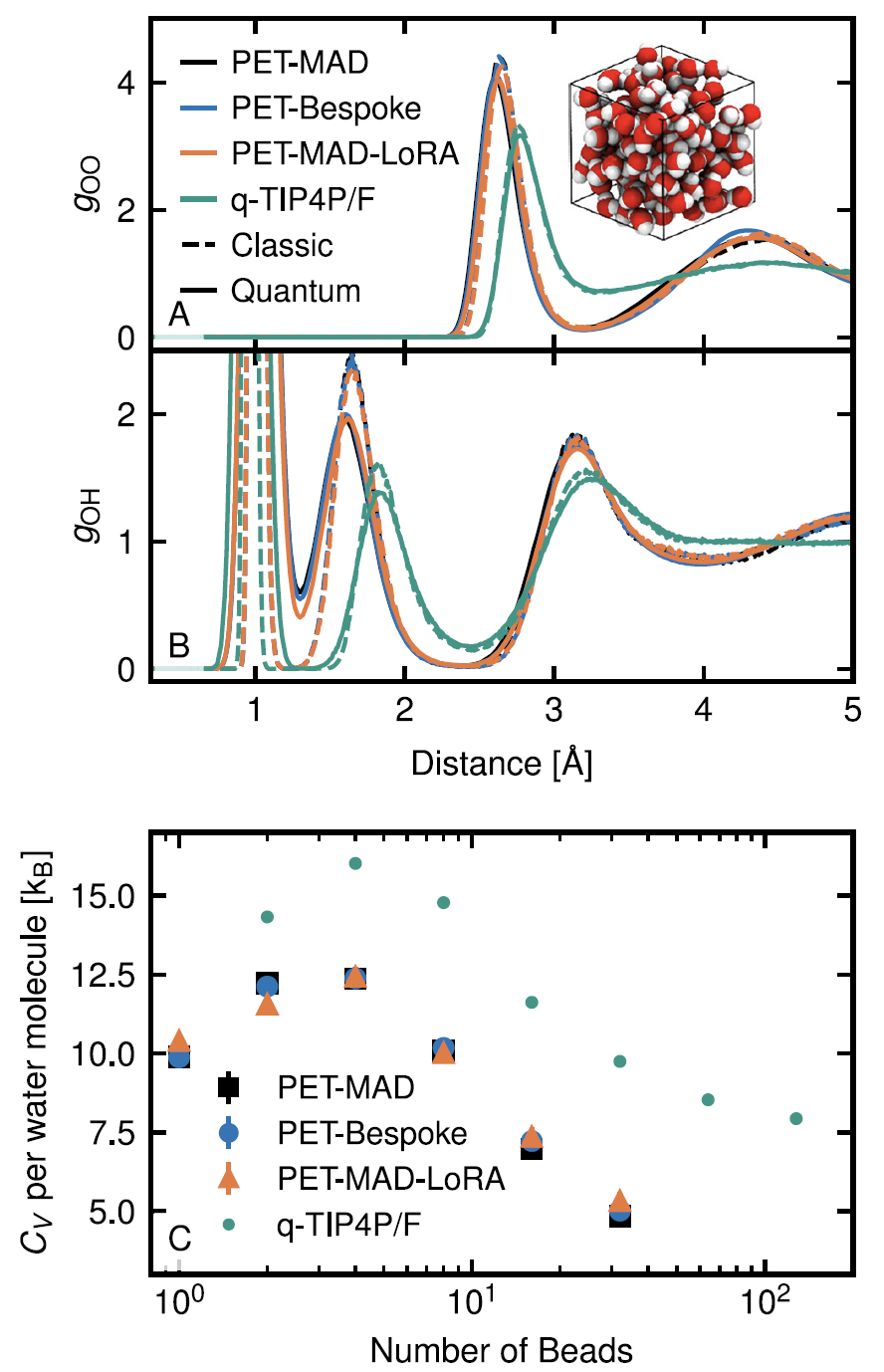
4. L'eau liquide et l'effet nucléaire quantique
Grâce à des simulations de dynamique moléculaire par intégrale de chemin, PET-MAD a permis de caractériser avec succès l'organisation structurale et les phénomènes de délocalisation des protons induits par les fluctuations quantiques des atomes d'hydrogène. Bien que limité par la précision de la fonctionnelle GGA utilisée, le système simulé présente des caractéristiques de « liquide surfondu » à température ambiante, et calcule avec précision la fonction de distribution radiale et la capacité thermique.Les résultats concordent parfaitement avec le modèle dédié PET-Bespoke, et son efficacité rend ces simulations quantiques, qui nécessitent une puissance de calcul importante, plus réalisables.
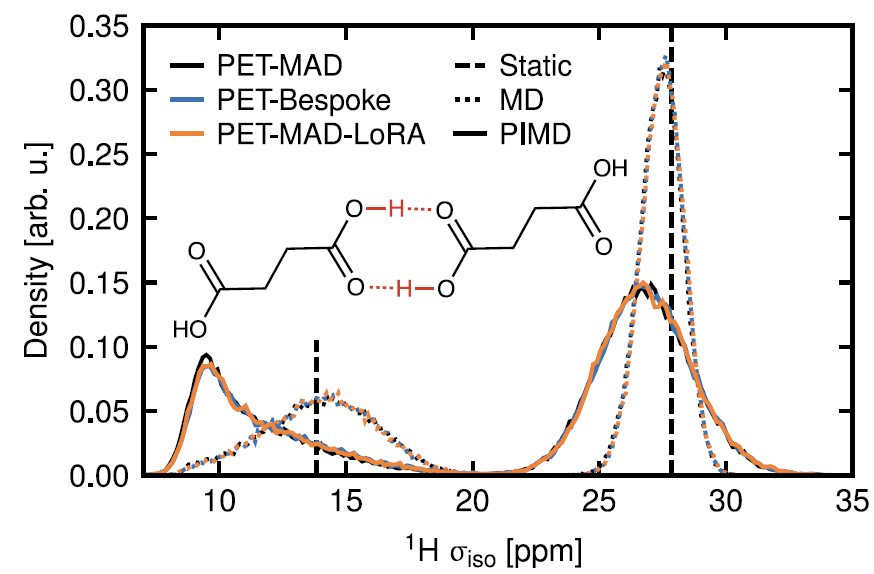
5. Cristaux moléculaires : étude RMN de l'acide succinique
Des simulations cinétiques pilotées par PET-MAD, combinées à un modèle de blindage chimique, ont permis de mettre en évidence l'influence du mouvement quantique nucléaire sur la distribution de la constante de blindage RMN du ¹H. Comme illustré dans la figure ci-dessous, la distribution du blindage des protons liés par liaisons hydrogène s'élargit et se déplace vers les basses énergies sous l'effet de l'échantillonnage quantique.Ceci est cohérent avec les conclusions du modèle dédié PET-Bespoke entraîné sur une DFT de haute précision, démontrant le potentiel de PET-MAD dans la prédiction de caractéristiques fonctionnelles complexes.
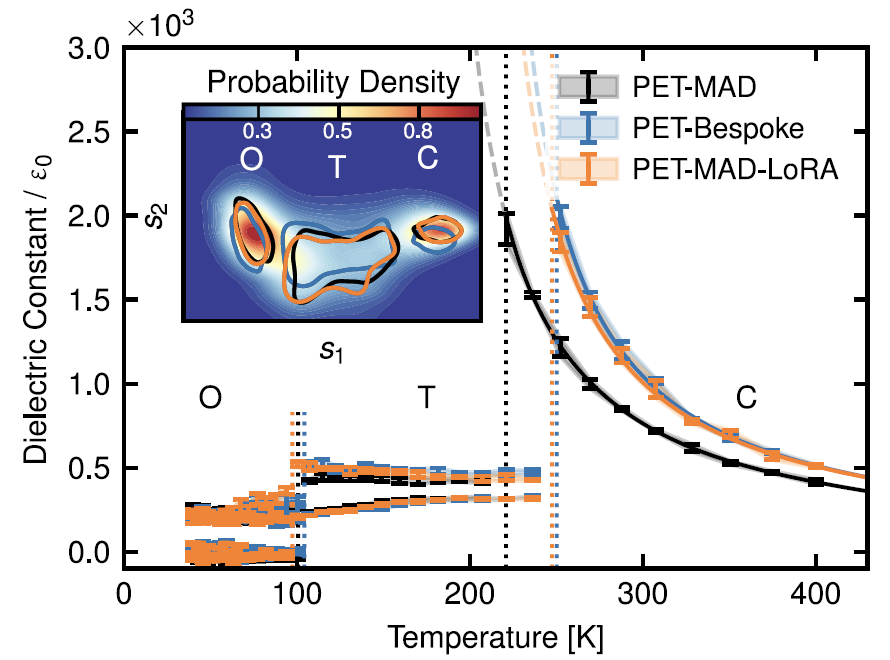
6. Matériaux ferroélectriques : Titanate de baryum (BaTiO₃)
Comme le montre la figure ci-dessous, dans les simulations de cellules flexibles réalisées dans la gamme de températures de 40 à 400 K, PET-MAD a reproduit avec précision la séquence caractéristique de transition de phase du matériau, de la structure rhomboédrique à la structure orthorhombique, puis tétragonale et enfin cubique. Malgré les limitations de la DFT…La température de transition de phase prédite est inférieure à la valeur expérimentale, mais son écart par rapport au résultat du modèle dédié PET-Bespoke est inférieur à 30 K.Dans le calcul de la constante diélectrique statique, elle a également correctement prédit la tendance d'une constante diélectrique élevée en phase cubique et d'une constante diélectrique faible en phase ferroélectrique.
Les tests de performance et les applications de systèmes multivariés démontrent tous deux que...PET-MAD peut atteindre une précision de simulation proche de celle des modèles dédiés PET-Bespoke avec très peu de données propriétaires, et prédire de manière fiable des propriétés telles que le transport d'ions, la transition de phase et la réaction de surface.Son réglage fin LoRA peut encore mieux se rapprocher des performances du modèle dédié PET-Bespoke, tandis que l'estimation d'erreur LLPR et le mode de prédiction de force double assurent l'efficacité et la robustesse des simulations complexes, ce qui en fait un outil de potentiel interatomique polyvalent, à la fois large et profond.
MLIPs : L’intégration industrie-université-recherche engendre un changement de paradigme dans la recherche et le développement des matériaux
Grâce à ses atouts majeurs que sont sa haute précision et son efficacité élevée, le domaine des MLIP (microscopies à impulsions liquides) s'impose de plus en plus comme un lien essentiel entre la pointe de la théorie de la chimie quantique et ses applications en ingénierie industrielle. Ces dernières années, la recherche s'est orientée de la vérification de la faisabilité des méthodes vers l'extension de leurs capacités.
Par exemple, une équipe de recherche de l'Université de Cambridge et de l'École polytechnique fédérale de Lausanne (EPFL) a combiné l'apprentissage actif avec des stratégies d'échantillonnage augmentées.Cela permet au modèle d'explorer de manière autonome les configurations clés de l'état de transition le long des voies de réaction chimique, permettant ainsi d'obtenir des prédictions précises de l'activité et de la sélectivité du catalyseur sans avoir besoin de données de réaction préalables exhaustives.Il surmonte efficacement les limitations des MLIP existants dans la description des processus dynamiques des réactions chimiques.
Titre de l'article : Aedes aegypti Argonaute 2 contrôle l'infection par les arbovirus et la mortalité de l'hôte
Lien vers l'article :
https://www.nature.com/articles/s41467-023-41370-y
Parallèlement, l'évaluation systématique et l'élucidation mécanistique des modèles existants s'approfondissent continuellement. L'équipe de l'Université de Californie a mené une évaluation exhaustive des principaux modèles MLIP à usage général, tels que M3GNet, CHGNet et MACE-MP-0.Il couvre un large éventail de propriétés des matériaux, notamment l'énergie de surface, l'énergie de formation des défauts, le spectre des phonons et la barrière de migration des ions.L'étude a révélé que ces modèles présentent généralement une sous-estimation systématique de l'énergie et de la force dans les tâches impliquant des états de haute énergie ou des configurations hors équilibre, ce qui ouvre la voie à de nouvelles améliorations dans les stratégies d'entraînement des modèles et la construction des données.
Titre de l'article : Adoucissement systématique des potentiels interatomiques universels d'apprentissage automatique
Lien vers l'article :
https://www.nature.com/articles/s41524-024-01500-6
En résumé, le développement des MLIP (Modèles d'Innovation à Grande Échelle) est entré dans une nouvelle phase, passant de l'innovation algorithmique aux approches axées sur la résolution de problèmes, et de l'évaluation fondamentale au développement approfondi de scénarios d'application. À l'avenir, à mesure que les défis de la modélisation multi-échelle seront relevés et que les modèles spécifiques à l'industrie gagneront en maturité, les MLIP devraient jouer un rôle plus central dans le système de R&D numérique, accélérant ainsi le renforcement mutuel des sciences des matériaux et de l'innovation industrielle.








