Command Palette
Search for a command to run...
Couvrant 200 Millions De Spectres De Masse Moléculaire, l'Académie Tchèque Des Sciences a Publié Le Modèle DreaMS Pour Créer Le Plus Grand Ensemble De Données De Spectrométrie De Masse Au Monde GeMS
Selon les statistiques, l'espace chimique naturel des petites molécules actuellement exploré par les humains représente moins de 10% de sa quantité totale, tandis que dans les expériences de métabolomique non ciblées, plus de 90% de spectres de masse sont devenus des « déchets de données » en raison du manque d'annotations fiables.
Dans cette bataille cruciale pour déchiffrer les molécules, le principal défi consiste à décrypter le langage complexe de la spectrométrie de masse en tandem (MS/MS). Outil de pointe pour l'analyse chimique moderne, le système chromatographie liquide-spectrométrie de masse en tandem (LC-MS/MS) permet une séparation efficace des molécules par chromatographie liquide, puis utilise la technologie de dissociation induite par collision pour générer des spectres de masse d'ions fragments. Ce processus est comparable au désassemblage d'une molécule et à l'analyse de son puzzle de fragments.
Cependant, les outils analytiques existants présentent des limites importantes pour reconstituer une image moléculaire complète :Même l’algorithme avancé SIRIUS dépend excessivement d’une bibliothèque spectrale limitée et de règles artificielles.Face à des molécules naturelles inconnues représentant plus de 801 TP3T au total, l'absence de bibliothèque à consulter est souvent un dilemme. Une étude publiée dans Nature Methods en 2023 a souligné que, dans la base de données métabolomique mondiale, seuls 21 spectres MS/MS de TP3T ont été annotés avec succès, les 981 TP3T restants constituant de véritables récifs en eaux profondes, entravant sérieusement la découverte de nouveaux médicaments et la recherche sur le diagnostic des maladies.
Afin de résoudre ce problème, une équipe de recherche de l'Institut de chimie organique et de biochimie de l'Académie tchèque des sciences s'est appuyée sur les avancées de la série GPT dans le domaine du langage et s'est engagée à créer un traducteur dédié aux spectres de masse. Les chercheurs ont exploité 700 millions de spectres MS/MS du Global Natural Products Social Molecular Network (GNPS), ont construit le plus grand ensemble de données de spectrométrie de masse de l'histoire, GeMS, et ont entraîné un modèle Transformer DreaMS avec 116 millions de paramètres. Ce modèle revient à laisser l'intelligence artificielle apprendre la « grammaire défectueuse » des molécules de toutes pièces. En prédisant les pics spectraux masqués et l'ordre de rétention chromatographique, ils ont découvert des schémas structuraux cachés dans des spectres de masse non marqués :Le vecteur de caractérisation à 1 024 dimensions qu'il génère peut refléter avec précision les similitudes structurelles entre les molécules et montrer une forte robustesse aux fluctuations du signal dans différentes conditions de spectrométrie de masse.
Les recherches montrent queLe DreaMS optimisé fonctionne bien dans une variété de tâches d'annotation de spectrométrie de masse.Y compris la prédiction de la similarité spectrale, des empreintes moléculaires, des propriétés chimiques et de la présence de fluor, qui surpassent tous les algorithmes traditionnels et les modèles d'apprentissage automatique récemment développés., DreaMS a intégré 201 millions de spectres pour construire un réseau super moléculaire couvrant les bactéries, les plantes et les métabolites humains,Elle a créé une « encyclopédie moléculaire » pour la communauté chimique qui peut être mise à jour en temps réel, fournissant des ressources extrêmement précieuses pour la recherche et les applications dans des domaines connexes.
Les résultats de recherche pertinents ont été publiés dans la revue de renommée internationale Nature Biotechnology sous le titre « Apprentissage auto-supervisé de représentations moléculaires à partir de millions de spectres de masse en tandem à l'aide de DreaMS ».

Adresse du document :
Autres articles sur les frontières de l'IA :
Adresse de téléchargement de l'ensemble de données de spectrométrie de masse chimique GeMS :
https://go.hyper.ai/IC2yw
Ensemble de données GeMS : 700 millions de spectres pour créer une base de données spectrales de masse
Les données de base de cette étude sont l’ensemble de données GeMS, extrait en profondeur du référentiel MassIVE GNPS, dont l’échelle et la qualité sont révolutionnaires dans le domaine de la métabolomique.
Adresse de téléchargement de l'ensemble de données de spectrométrie de masse chimique GeMS :
https://go.hyper.ai/IC2yw
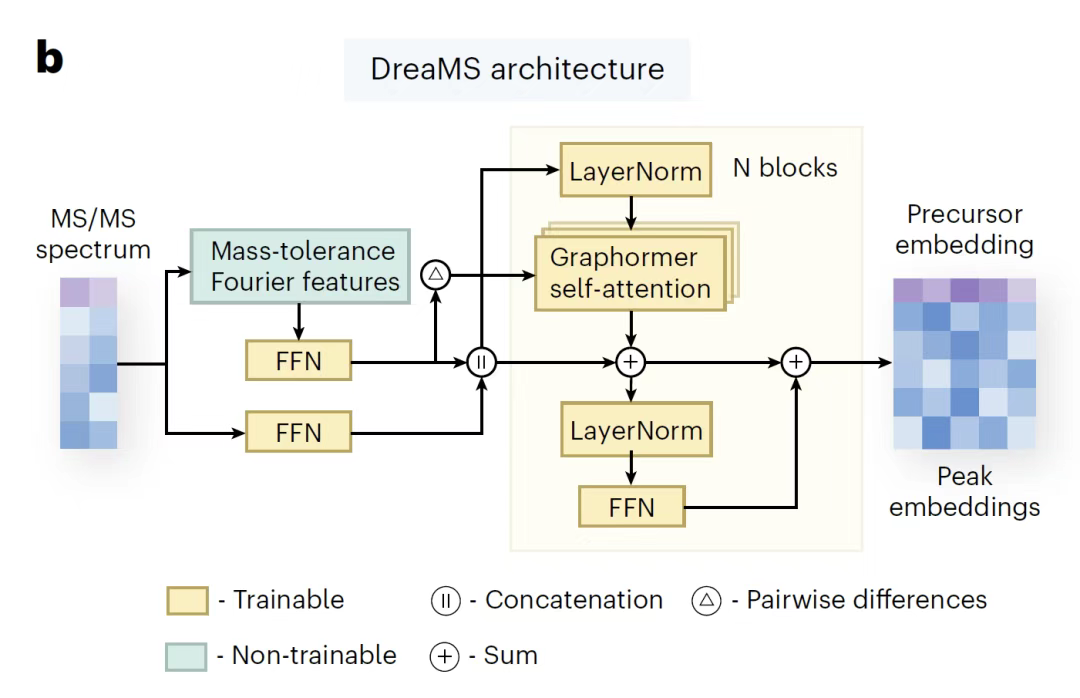
Comme le montre la figure ci-dessous,L'équipe de recherche a intégré 250 000 données expérimentales LC-MS/MS couvrant les domaines biologiques et environnementaux, en a extrait environ 700 millions de spectres MS/MS et les a divisés en trois sous-ensembles : GeMS-A, GeMS-B et GeMS-C grâce à des algorithmes de contrôle qualité stricts.Parmi eux, GeMS-A collecte principalement des spectres avec le spectromètre de masse Orbitrap 97%, représentant le plus haut standard de qualité ; GeMS-C intègre les spectres Orbitrap 52% et QTOF 41%, élargissant considérablement l'échelle des données tout en garantissant une certaine qualité. Cette conception hiérarchique préserve non seulement la fiabilité des données des instruments de haute précision, mais couvre également un plus large éventail de sources technologiques de spectrométrie de masse grâce à des sous-ensembles plus complets, garantissant ainsi la diversité des jeux de données.
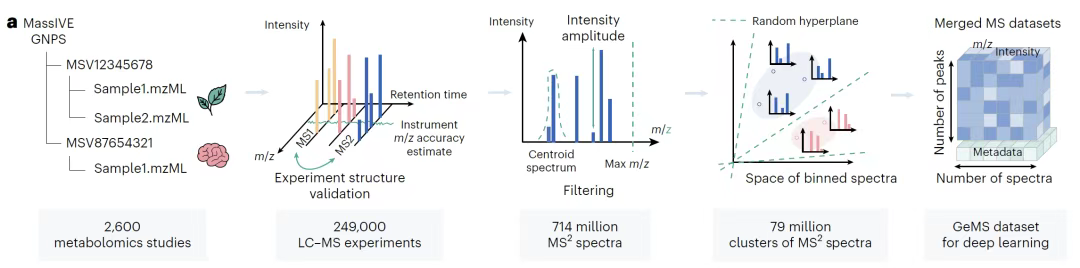
Pour résoudre le problème de redondance des données à grande échelle, l'équipe de recherche a utilisé l'algorithme de hachage sensible à la localité (LSH) pour regrouper efficacement les spectres similaires et a généré neuf variantes en limitant le nombre de spectres dans le cluster, optimisant ainsi l'efficacité des calculs tout en préservant la représentativité des données. L'ensemble de données GeMS a finalement été stocké au format binaire compact HDF5.Convertir le spectre brut en un tenseur numérique de dimension fixe,Il élimine le goulot d'étranglement des bibliothèques spectrales traditionnelles : comme le montre la figure ci-dessous, son volume de données est plusieurs fois supérieur à celui des bibliothèques existantes et sa structure est hautement standardisée, offrant ainsi des ressources d'apprentissage sans précédent pour les modèles d'apprentissage profond. Ces caractéristiques font de GeMS le premier ensemble de données de spectrométrie de masse à très grande échelle adapté à l'apprentissage non supervisé/auto-supervisé. Il pose non seulement les bases du pré-apprentissage du modèle DreaMS, mais fournit également un support de données précis et complet pour les analyses ultérieures de similarité spectrale, la caractérisation de la structure moléculaire et d'autres tâches grâce à une stratification de qualité et à l'optimisation du format, favorisant ainsi la recherche en métabolomique, passant du modèle traditionnel reposant sur des bibliothèques de référence limitées au paradigme d'analyse intelligente basé sur des spectres bruts massifs.
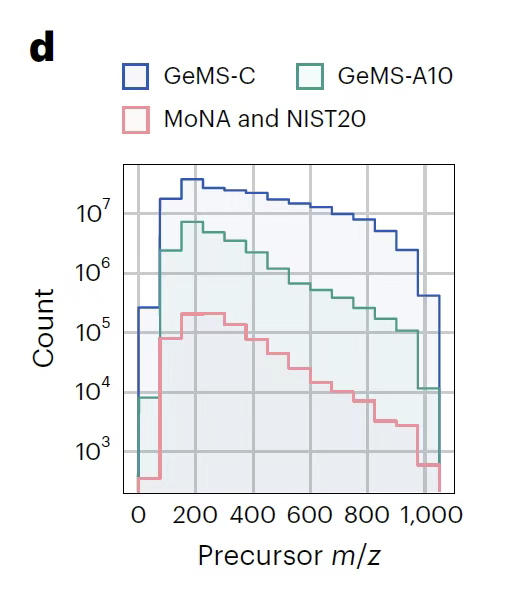
Modèle DreaMS : un nouveau paradigme pour l'analyse par spectrométrie de masse basé sur le transformateur auto-supervisé
Basé sur l'ensemble de données GeMS, le modèle DreaMS vise à extraire des représentations moléculaires à partir de spectres MS/MS non annotés via un apprentissage auto-supervisé.Ce modèle s'appuie sur l'architecture BERT dans le traitement du langage naturel et a été le pionnier d'un paradigme d'apprentissage auto-supervisé dans le domaine de la spectrométrie de masse des petites molécules.Sa conception principale comprend deux objectifs de formation : l'un consiste à masquer aléatoirement le rapport masse/charge (m/z) de 30% dans le spectre proportionnellement à l'intensité, et à entraîner le modèle à reconstruire les pics masqués, tout en introduisant des « étiquettes d'ions parents » pour agréger les informations au niveau spectral (similaire à la représentation au niveau des phrases des modèles de langage) ; l'autre consiste à apprendre à prédire l'ordre d'élution chromatographique à travers des paires spectrales de la même expérience LC-MS/MS, et à renforcer la relation intrinsèque entre la structure moléculaire et les règles d'élution des pics.
En termes d’architecture du modèle, comme le montre la figure ci-dessous,DreaMS est basé sur un encodeur Transformer à 7 couches équipé d'un mécanisme d'auto-attention à 8 têtes, qui peut générer un vecteur de représentation de 1 024 dimensions.Pour les données de rapport masse/charge à haute résolution, le modèle utilise la technologie de prétraitement des caractéristiques de Fourier pour décomposer les valeurs de masse continues en composantes de fréquence sinus/cosinus, capturer les détails des parties entières et à virgule flottante, et associer ultérieurement les prédictions de composition des éléments via un réseau à propagation directe ; la valeur d'intensité est traitée par un réseau superficiel et concaténée avec les caractéristiques de Fourier en entrée du transformateur. De plus,DreaMS introduit explicitement les différences de caractéristiques de Fourier de toutes les paires de pics dans la tête d'auto-attention (en empruntant à l'architecture Graphormer).Modélisez directement la relation de perte neutre, en évitant un étiquetage supplémentaire ou des calculs complexes.
Cette étude a utilisé une technique de sondage linéaire pour évaluer les changements dans les représentations acquises pendant la phase de formation.Premièrement, au cours du processus de formation, le modèle de régression logistique basé sur le vecteur d'intégration de l'ion parent peut prédire progressivement l'empreinte digitale de liaison MACCS, indiquant que le modèle apprend les informations sur les fragments moléculaires en auto-supervision ; deuxièmement, l'analyse de la tête d'attention montre que le modèle donne la priorité aux pics caractéristiques représentant la structure moléculaire plutôt qu'au bruit ; enfin, les résultats du clustering de l'espace de caractérisation montrent que même les spectres dans différentes conditions d'ionisation peuvent être distribués linéairement en fonction de la structure moléculaire, vérifiant sa capacité à capturer les caractéristiques structurelles.
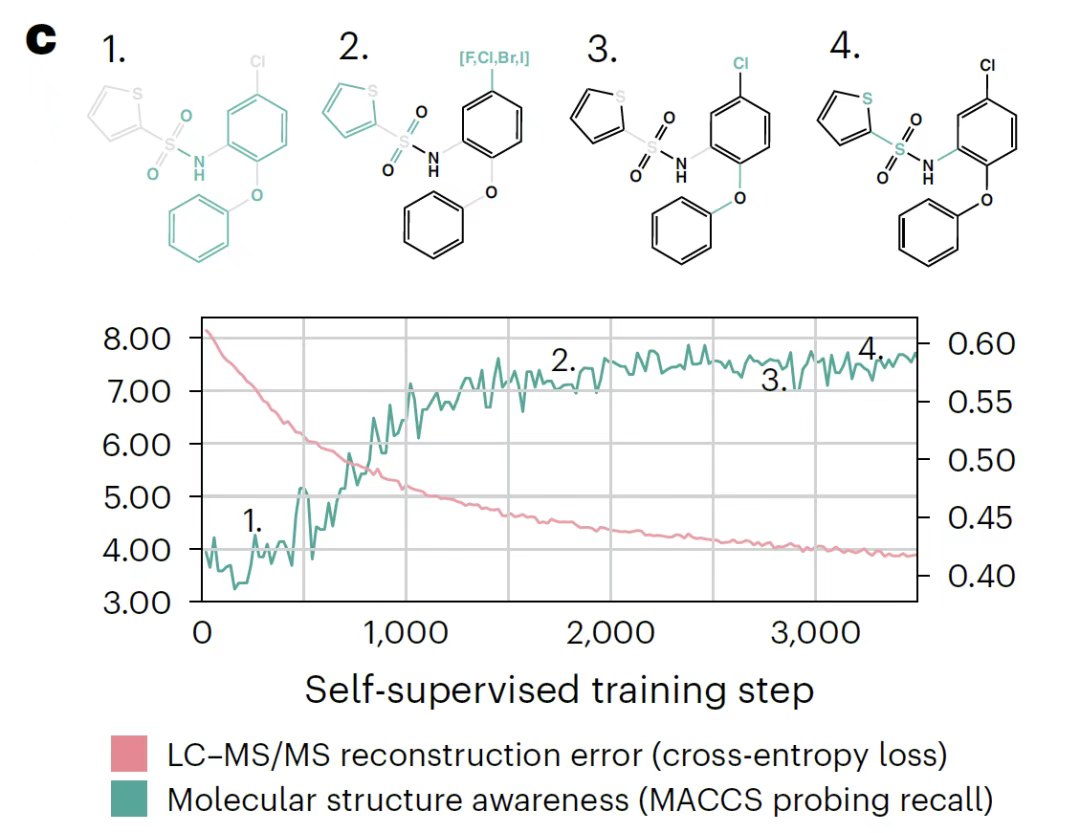
Migration inter-tâches des modèles DreaMS : analyse par spectrométrie de masse de l'analyse de molécule unique à l'interconnexion complète du métabolome
Premier modèle d'analyse par spectrométrie de masse basé sur l'apprentissage auto-supervisé, le modèle DreaMS a démontré des avantages significatifs en termes de migration inter-tâches. L'équipe de recherche l'a adapté à quatre tâches principales :
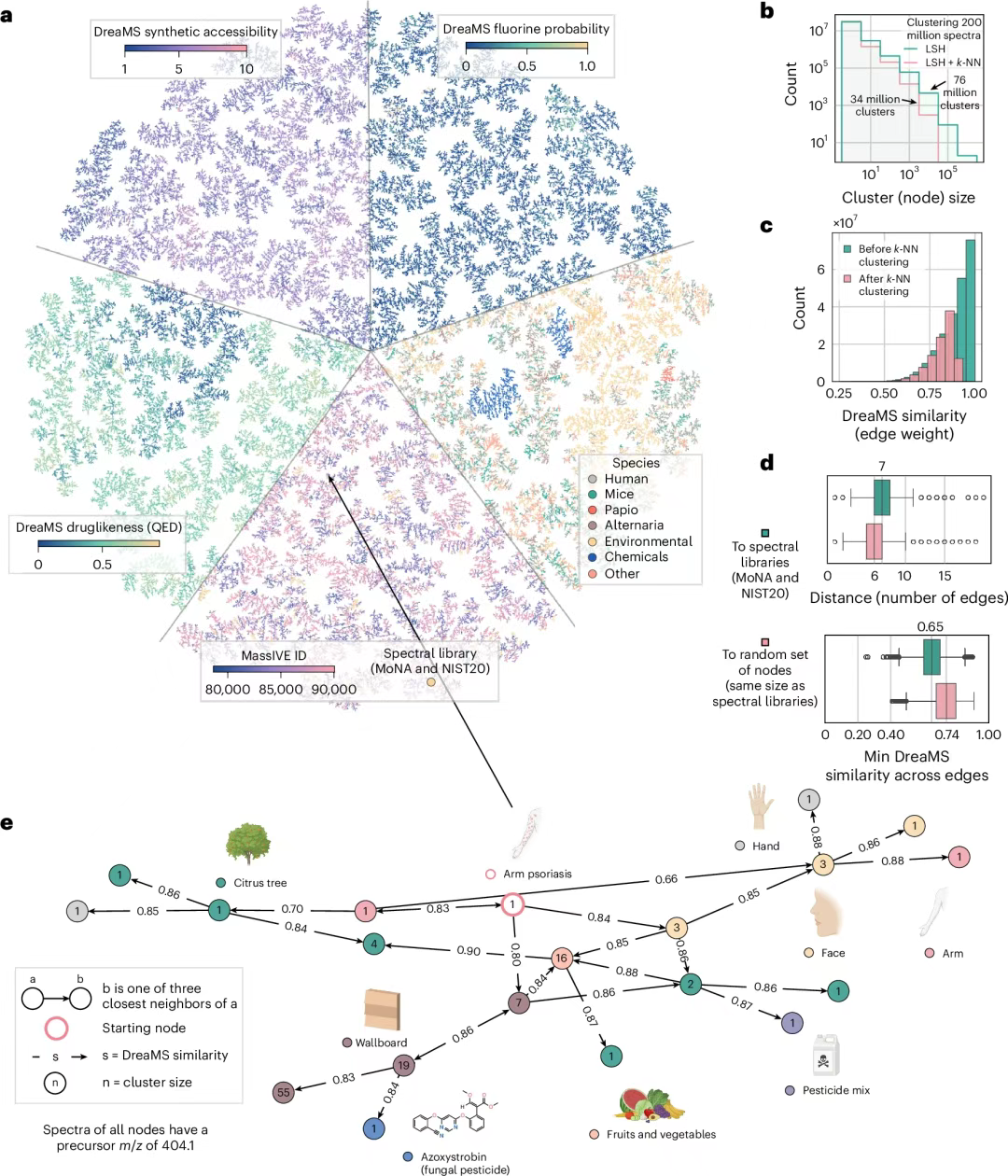
Dans l'analyse de similarité spectrale,Comme le montre la figure ci-dessous, le modèle réalise d'abord une correspondance à échantillon nul grâce à une caractérisation auto-supervisée. La corrélation entre la similarité cosinusoïdale de l'espace d'inclusion et la similarité de la structure moléculaire (comme le coefficient de Tanimoto) dépasse l'algorithme supervisé MS2DeepScore, qui repose sur l'apprentissage de données étiquetées. Compte tenu de la limitation liée à l'insensibilité des échantillons nuls aux différences subtiles de structure moléculaire, un triplet d'exemples complexes, comprenant des spectres de référence, des échantillons positifs de la même molécule et des échantillons négatifs de masse similaire, est conçu pour la comparaison et l'affinage. Ainsi, lors de la tâche de récupération avec un écart de masse du précurseur inférieur à 10 ppm,Le DreaMS affiné surpasse considérablement 44 mesures de similarité traditionnelles.De plus, les résultats d'intégration sont plus robustes aux différences entre les instruments de spectrométrie de masse, et l'analyse UMAP montre que son espace de représentation est strictement regroupé selon les formules chimiques moléculaires et les motifs structurels.
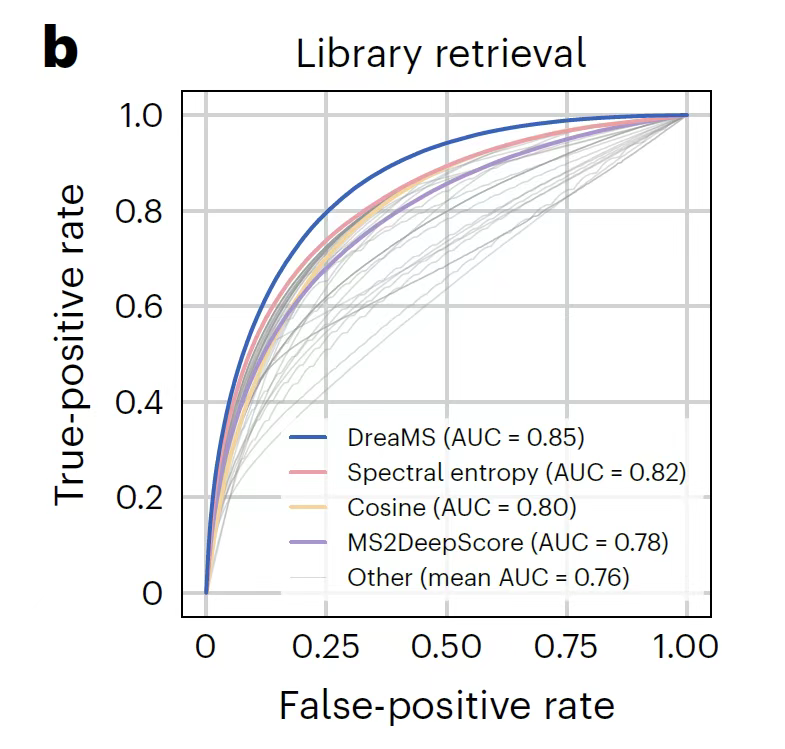
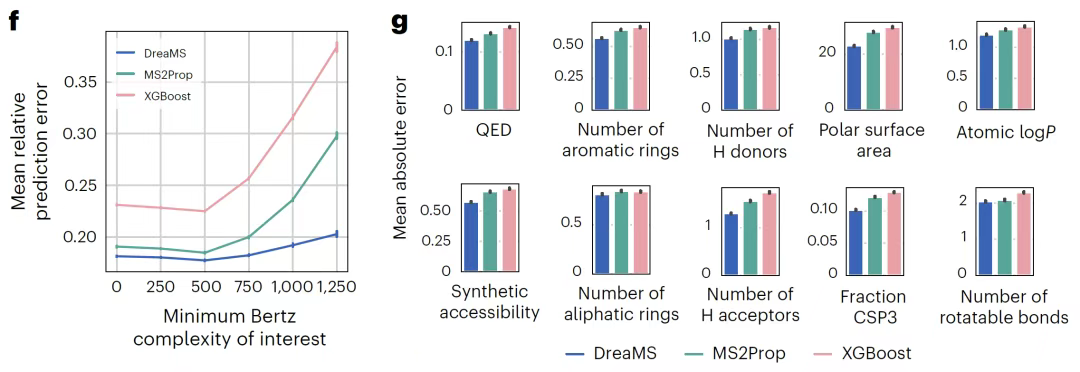
Dans la tâche de prédiction d'empreintes digitales moléculaires,Comme le montre la figure ci-dessous, DreaMS s'affranchit du processus complexe des méthodes traditionnelles qui reposent sur l'attribution de formules chimiques ou la génération d'arbres de fragments. Un seul passage direct permet de prédire directement les empreintes de Morgan à partir de spectres bruts. Les performances de recherche dans la base de données PubChem sont comparables à celles du modèle d'apprentissage profond MIST, qui repose sur l'annotation des formules chimiques de pic, mais il omet les étapes intermédiaires gourmandes en calcul. Pour la prédiction des propriétés chimiques liées aux produits pharmaceutiques, le modèle génère les cinq paramètres de la règle de Lipinski, la complexité moléculaire de Bertz et d'autres indicateurs par ajustement fin.Il a obtenu les meilleures performances actuelles dans les scénarios de criblage de médicaments à grande échelle et de recherche de biomarqueurs extraterrestres.
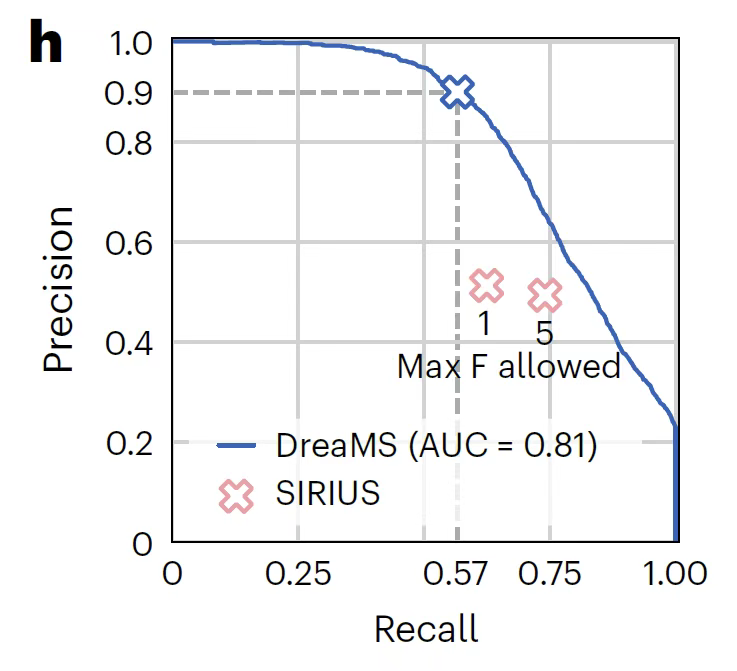
Dans la tâche la plus difficile de détection de molécules fluorées,Comme le montre la figure ci-dessous, DreaMS atteint une précision de 0,91 et un rappel de 0,57 grâce à un modèle de prédiction probabiliste.C'est bien supérieur à l'algorithme SIRIUS qui s'appuie sur la recherche de combinaison de règles de fragmentation et a une précision de seulement 0,51.En particulier, il présente une forte capacité de généralisation dans la détection de molécules aux structures nouvelles, fournissant un outil clé pour le développement de médicaments liés au fluorure et la surveillance environnementale.
Grâce à une grande efficacité de calcul (l'intégration d'un million de spectres ne prend qu'une heure sur le GPU NVIDIA A100), comme le montre la figure ci-dessous, l'équipe de recherche a construit un graphe DreaMS contenant 201 millions de spectres de masse et généré un graphe des trois plus proches voisins (3-NN) de 34 millions de nœuds grâce au clustering par hachage sensible local. La similarité des arêtes de 67% est supérieure à 0,8, et 99,71 nœuds TP3T forment un seul composant connexe. L'analyse du plus court chemin montre qu'elle peut connecter n'importe quel spectre à une entrée de bibliothèque connue en 6 étapes.
Dans une étude métabolomique du psoriasis du bras,Comme le montre la figure e ci-dessous, la carte révèle l'association potentielle entre la maladie et le fongicide pyraclostrobine grâce à la connectivité spectrale. Cette voie d'association implique des sources d'exposition environnementales telles que les aliments contaminés et les arbres traités, offrant une nouvelle perspective, basée sur les données, pour l'exploration des causes de maladies complexes. Cette capacité à annoter avec précision une seule tâche pour déduire l'ensemble du réseau de bibliothèques marque une nouvelle ère dans laquelle la technologie d'analyse par spectrométrie de masse est passée du « décodage de molécule unique » à « l'interconnexion complète du métabolome ».
La collaboration entre l'industrie, l'université et la recherche stimule l'innovation dans la technologie de la spectrométrie de masse
Dans le domaine de l’analyse par spectrométrie de masse de petites molécules et de la recherche métabolomique, les universités et les entreprises du monde entier utilisent des technologies innovantes pour promouvoir des percées dans ce domaine.
En matière de recherche universitaire, la technologie d'analyse de données massives multi-omiques assistée par l'IA, développée par le laboratoire de Hu Zeping à l'Université Tsinghua en Chine, combinée à des méthodes métabolomiques de haute précision, a permis de révéler le mécanisme d'interaction métabolique entre les neurones et les cellules cancéreuses dans le microenvironnement tumoral et de découvrir des voies de régulation des neurotransmetteurs pouvant servir de cibles thérapeutiques. Ses résultats ont été maintes fois analysés par la revue Nature. Le système expert de caractérisation CataAI, développé par l'Institut de physique chimique de Dalian de l'Académie chinoise des sciences,En intégrant la technologie d’apprentissage profond dans le processus d’analyse des données de spectrométrie de masse et en utilisant des bases de données auto-construites et de nouveaux algorithmes, nous avons obtenu des recommandations intelligentes allant des spectres de masse aux structures moléculaires.Un modèle de réseau neuronal en deux étapes a été développé pour les données de caractérisation complexes des matériaux catalytiques énergétiques.
La plateforme Global Natural Products Social Molecular Network (GNPS) de l'Université de Californie à San Diego (UCSD)En tant que source de l'ensemble de données de base GeMS du modèle DreaMS étudié dans cet article, il continue de promouvoir le partage et l'intégration des données de spectrométrie de masse interinstitutionnelles.Ses dernières recherches ont établi une méthode d’analyse métabolomique du microbiome intestinal à haut débit en comparant les systèmes de solvants à base d’éthanol et de méthanol, fournissant un processus standardisé pour analyser les mécanismes d’interaction hôte-microbe.
Dans le cadre des pratiques d'innovation d'entreprise, la société américaine Agilent a lancé une nouvelle génération de systèmes de détection de la qualité des liquides tels que la série Pro iQ, qui présentent d'excellentes performances et une sensibilité excellentes et sont idéales pour la surveillance de molécules biologiques complexes et la détection d'impuretés.Sa gamme de masse est étendue à m/z 2–3000 et sa sensibilité est améliorée par la technologie Agilent Jet Stream (AJS).Il prend en charge la détection de routine et de traces de petites molécules et de macromolécules, offrant ainsi des moyens techniques révolutionnaires pour la supervision de la sécurité alimentaire. S'appuyant sur la technologie de chromatographie liquide couplée à la spectrométrie de masse en tandem, l'entreprise chinoise Kailaipu Technology a développé de manière indépendante plus de 20 kits cliniques de spectrométrie de masse, couvrant plus de 300 éléments de détection. Parmi eux, les réactifs de détection des métabolites des catécholamines dans le sang et l'urine ont été inclus dans le consensus d'experts de la Société d'endocrinologie de l'Association médicale chinoise et sont devenus la référence clinique.
De manière générale, le domaine actuel de l'analyse par spectrométrie de masse des petites molécules et de la recherche en métabolomique connaît une innovation technologique portée par les universités et les entreprises. Ces innovations non seulement approfondissent la compréhension humaine de la complexité des systèmes biologiques en théorie, mais présentent également un fort potentiel d'applications pratiques, du diagnostic précoce du cancer à la prédiction du pronostic des maladies cardiovasculaires, de la recherche et du développement de matériaux catalytiques à la surveillance de la sécurité alimentaire. Cette révolution, déclenchée par la résonance de l'innovation algorithmique et de la science expérimentale, pourrait reconstruire complètement l'écologie de la chaîne, de la recherche fondamentale à l'application clinique, et avoir des répercussions plus profondes dans les domaines connexes.
Enfin, je voudrais recommander un événement à tous : HyperAI organisera le 7e salon Meet AI Compiler Technology à Pékin le 5 juillet.Nous avons la chance d'avoir invité de nombreux experts seniors d'AMD, de l'Université de Pékin, de Muxi Integrated Circuit, etc. Bienvenue à tous pour cliquer sur le lien ci-dessous pour vous inscrire~
https://www.huodongxing.com/event/1810501012111
Articles de référence :
1.https://mp.weixin.qq.com/s/1QUjLMtj_6ui9T0gbuZtrA
2.https://dicp.cas.cn/xwdt/ttxw/202411/t20241107_7435521.html
3.https://ccms-ucsd.github.io/GNPSDocumentation/
4.https://mp.weixin.qq.com/s/Wgh2w0G76koqc9AY0PBHcg








