Command Palette
Search for a command to run...
Les Dernières Recherches De l'équipe De David Baker Utilisent Des Modèles De Génération De Séquences Protéiques Pour Obtenir Une Conception De Gènes Chevauchants Avec Un Taux De Réussite Très élevé.
En 1977, le biochimiste britannique Frederick Sanger a découvert un phénomène qui a bouleversé la cognition pour la première fois en analysant le génome du bactériophage ΦX174 : la longueur totale des protéines codées par cette molécule d'ADN de 5,4 kb dépassait de loin sa limite de capacité physique. Les résultats du séquençage ont révélé queDeux paires de gènes partagent la même région d'ADN via des cadres de lecture différents - ce phénomène est appelé gènes qui se chevauchent (OLG) et est extrêmement courant dans le monde viral.Par exemple, dans le génome de 3,2 kb du virus de l’hépatite B, la région 50% est couverte par plusieurs paires de gènes qui se chevauchent, et plus de la moitié des virus connus contiennent au moins un OLG.
Cette conception du génome peu intuitive cache la sagesse de survie du virus : lorsque les virus se disputent un espace limité dans les cellules hôtes, OLG utilise la stratégie de « l'empilement de gènes » pour permettre à un seul nucléotide de participer au codage de deux codons en même temps, réalisant ainsi une superposition fonctionnelle dans une séquence compacte. La découverte de l’équipe Sanger a donné lieu à des recherches connexes. Des études ultérieures ont montré que les protéines codées par OLG présentent souvent une dégénérescence de séquence élevée et que leur tolérance à la séquence d'acides aminés permet à deux protéines fonctionnelles de coexister sur la même chaîne d'ADN. Plus important encore, même les protéines qui doivent former une structure tridimensionnelle claire peuvent atteindre une compatibilité de repliement dans différents cadres de lecture grâce à l’agencement des séquences.
Cependant, la question fondamentale demeure toujours : dans le cadre du code génétique standard, la dégénérescence des séquences d’acides aminés peut-elle permettre le repliement de paires de protéines fonctionnelles arbitraires dans des structures qui se chevauchent ? Lorsque les nucléotides doivent prendre en compte le double codage, l’espace de séquence pour le repliement des protéines est-il sévèrement restreint ?
L'équipe de David Baker de l'Université de Washington a récemment utilisé des modèles génératifs avancés pour mener des recherches sur la conception synthétique d'OLG et vérifier sa faisabilité d'un point de vue technique.L'équipe de recherche a conçu des séquences superposées pour deux familles de protéines afin de coder des structures protéiques conçues de novo hautement ordonnées. La simulation par ordinateur et la vérification expérimentale ont toutes deux montré un taux de réussite très élevé : sous des contraintes de chevauchement, les cadres de lecture alternatifs peuvent non seulement accueillir un pliage tridimensionnel clair, mais leur stabilité structurelle et leur intégrité fonctionnelle sont également comparables à celles des séquences non chevauchantes.
Les résultats de recherche pertinents ont été publiés sous forme de pré-impression sur bioRxiv sous le titre « Conception de gènes chevauchants à l'aide de modèles génératifs profonds de séquences protéiques ».

Adresse du document :
https://doi.org/10.1101/2025.05.06.652464
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : Intégration de ressources de données multidimensionnelles et de méthodes d'analyse
Afin d'analyser la plasticité du code génétique et son application dans la conception de protéines, cette étude intègre des ressources de données multidimensionnelles et des méthodes d'analyse pour construire une chaîne de recherche complète allant de la conception théorique à la vérification expérimentale.
Dans les études de randomisation du code génétique,L’étude a généré 1 000 combinaisons alternatives de codons basées sur des stratégies de permutation d’acides aminés et de mélange de codons.Cet ensemble de données garantit la diversité et l'uniformité des échantillons grâce à une conception d'algorithme claire, fournissant une référence statistique pour évaluer l'impact fonctionnel des réarrangements de codons.
Dans le même temps, l'étude a sélectionné 3 protéines cibles de structure secondaire représentatives et construit 9 combinaisons appariées, ce qui a permis de standardiser les conditions expérimentales sous le principe de contrôle des variables et de relier efficacement l'analyse de corrélation entre la variation du code génétique et la fonction de la structure des protéines.
Dans la phase d'analyse de la séquence du domaine protéique, l'étude a extrait les séquences de départ de la base de données Pfam 37.0, échantillonné au hasard des sous-régions de 100 acides aminés de longueur et utilisé le modèle de Markov pour générer des séquences de protéines synthétiques qui conservaient la distribution k-mer.Cette méthode combine le criblage bioinformatique et la modélisation statistique, qui non seulement conserve les caractéristiques de séquence des protéines naturelles, mais crée également des échantillons de contrôle en introduisant des variables aléatoires contrôlables.Il fournit un ensemble de données innovant qui combine des propriétés naturelles avec des caractéristiques conçues artificiellement pour une analyse ultérieure.
Dans l'analyse d'intégration du modèle de langage protéique, les chercheurs ont extrait les caractéristiques de la couche cachée d'ESM2, ESM3 et ProstT5 et les ont projetées dans un espace bidimensionnel via l'algorithme UMAP après calcul de la moyenne de position. En définissant avec précision des paramètres tels que n_neighbors = 15, les caractéristiques de séquence de grande dimension sont transformées en cartes topologiques intuitives.Tout en préservant la structure de similarité de séquence, il fournit un cadre de visualisation unifié pour la comparaison entre modèles.Il démontre la combinaison de pointe de la biologie computationnelle et de la visualisation des données.
Au cours de la phase de vérification expérimentale,Les chercheurs ont cloné et recombiné 192 gènes qui se chevauchent pour générer 384 variantes de protéines à cadre décalé.Les paramètres clés ont été strictement contrôlés dans l'expérience : 20 heures de culture à 37°C ont assuré la stabilité du système d'expression d'E. coli, et un schéma de renaturation par gradient de chlorhydrate de guanidine 6M a assuré le repliement correct de la protéine du corps d'inclusion. Ce contrôle quantitatif de l’ensemble du processus, de la conception moléculaire à la purification et à la caractérisation, améliore non seulement la reproductibilité des conclusions de la recherche, mais fournit également un paradigme expérimental standardisé pour l’ingénierie des protéines.
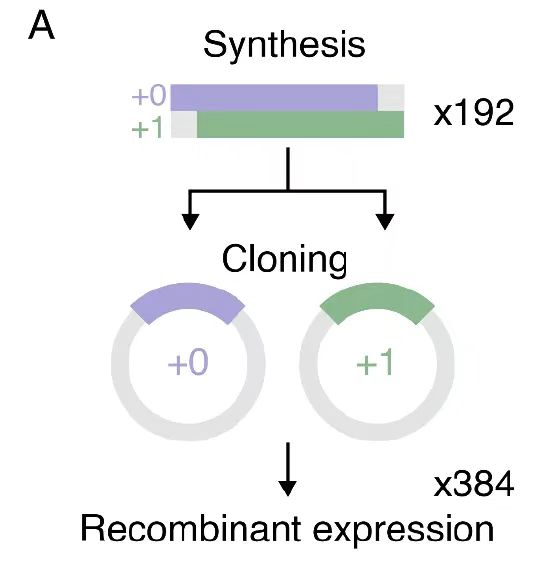
Conception OLG basée sur un modèle génératif : une méthode d'optimisation de synchronisation de séquence compatible multi-framework
Cette étude a développé un algorithme de calcul qui aborde efficacement le problème des contraintes d'espace de séquence causées par l'interdépendance des cadres de codage dans la conception de gènes qui se chevauchent (OLG), et a permis d'optimiser simultanément l'adaptabilité de deux séquences protéiques.
Au niveau de la conception de l’algorithme, l’étude a intégré des modèles génératifs tels que EvoDiff-MSA et ProteinMPNN.Le premier est basé sur l'architecture MSA Transformer et peut générer des séquences de conception basées sur l'alignement de séquences multiples de protéines cibles (MSA) grâce à un entraînement de cible par diffusion autorégressive ; ce dernier, en tant que modèle de génération conditionnelle structurelle, peut concevoir des séquences protéiques correspondantes lorsqu'une structure tridimensionnelle est donnée. Les deux modèles ont utilisé des stratégies de masquage position par position et d'échantillonnage contraint pour générer des bibliothèques de séquences superposées couvrant une variété de décalages et d'agencements de cadres.
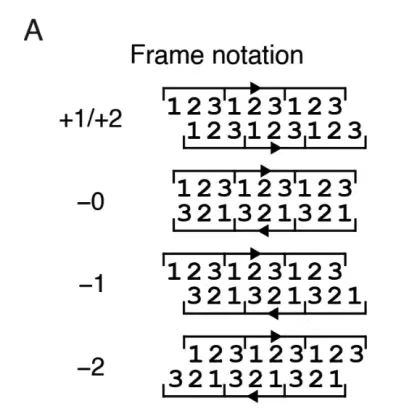
Comme le montre la figure A ci-dessous, cette étude a proposé une stratégie d’échantillonnage itératif image par image pour les contraintes de phase de cinq cadres de lecture variables (+1, +2, -0, -1 et -2).
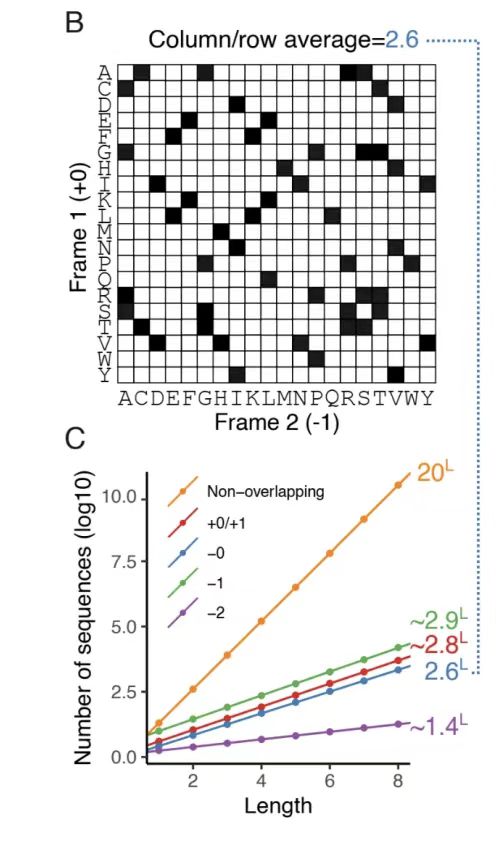
Comme le montre la figure B ci-dessous, en analysant la matrice de compatibilité des acides aminés du cadre -0, il a été constaté qu'il existe en moyenne 2,6 choix d'acides aminés compatibles à une seule position dans le cadre de référence, formant 52ⁿ (n est la longueur de la séquence) paires de séquences potentiellement chevauchantes, mettant en évidence l'espace de conception apporté par la dégénérescence du code génétique. Les degrés de liberté d’autres cadres ont été quantifiés à l’aide de l’approximation de Monte Carlo, comme le montre la figure C ci-dessous. Les résultats montrent que les cadres +1 et -1 ont des degrés de liberté plus élevés (environ 2,8 et 2,9, respectivement), tandis que le cadre -2 a des degrés de liberté considérablement limités (environ 1,4) en raison de la faible efficacité de la dégénérescence des codons.
Enfin, comme le montre la figure D ci-dessous, l'algorithme analyse systématiquement les positions de séquence (ordre d'analyse) et met à jour dynamiquement la matrice de probabilité conjointe à chaque analyse en combinaison avec les contraintes d'acides aminés adjacents.Après plusieurs séries d'itérations, les paires de séquences superposées générées sont assurées de respecter la compatibilité du framework.Cette stratégie peut être étendue à des cadres complexes avec des décalages de phase, optimisant la qualité de conception en biaisant l'ordre de balayage et en fournissant des contraintes clés pour le décodage itératif du modèle génératif.
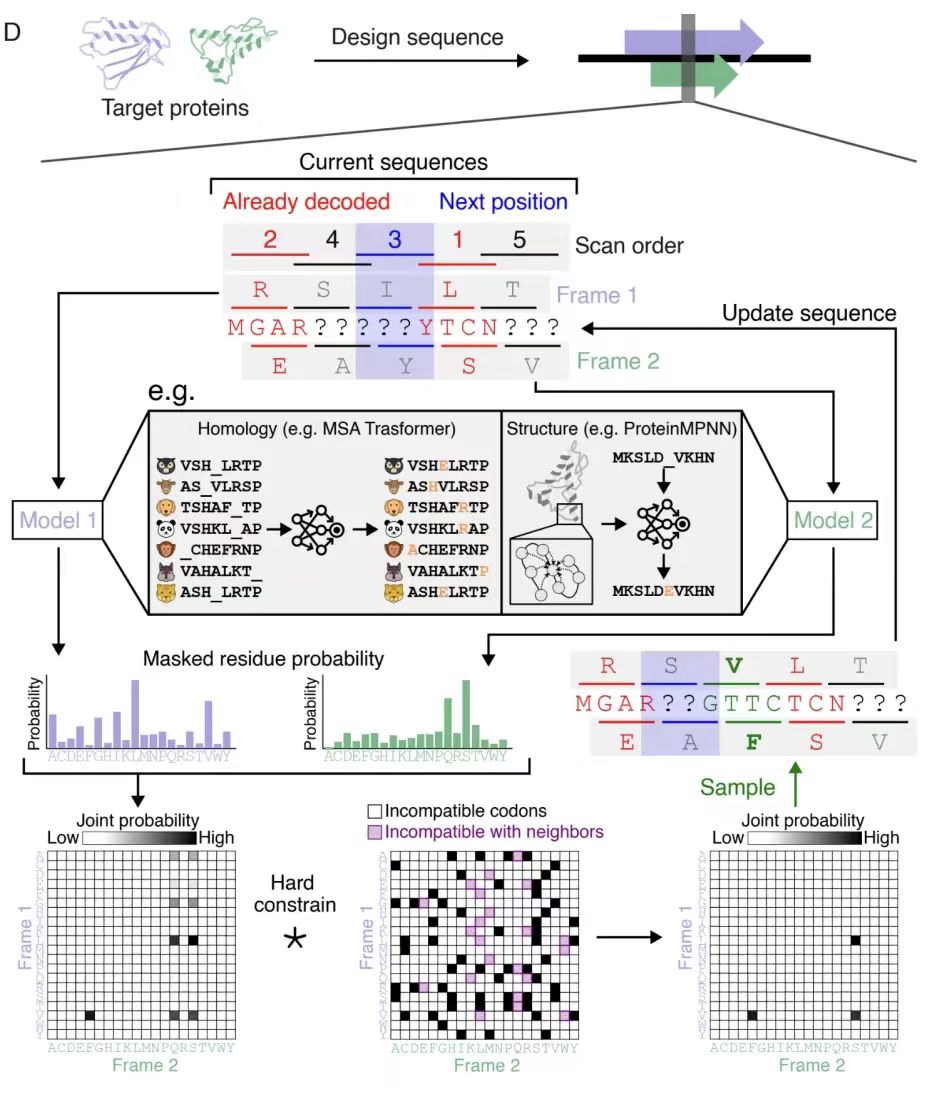
Au-delà de la limitation du modèle naturel : génération efficace de OLG synthétiques de paires de protéines arbitraires
La conception expérimentale couvre plusieurs directions, notamment l'évaluation de la conception OLG basée sur l'homologie, l'analyse de faisabilité du chevauchement des structures de chaîne principale de protéines hautement ordonnées, les études d'accessibilité évolutive des séquences OLG et la vérification expérimentale.
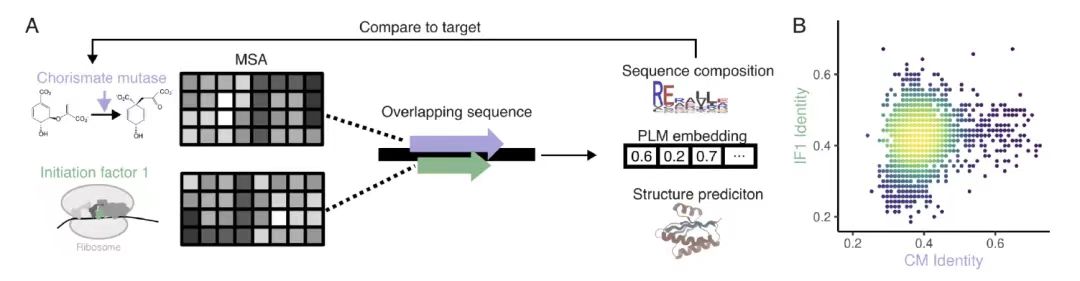
Dans l'évaluation de la conception OLG basée sur l'homologie,Comme le montre la figure A ci-dessous, l'équipe de recherche a sélectionné la shikimate mutase bactérienne (CM) et le facteur d'initiation de la traduction 1 (IF1) comme cibles, a utilisé le modèle de génération EvoDiff-MSA et a utilisé l'alignement de séquences multiples (MSA) comme contexte conditionnel pour générer 3 307 conceptions de séquences complètement superposées grâce au masquage position par position et à l'échantillonnage contraint.
Comme le montre la figure B ci-dessous, bien que l'homologie entre la séquence conçue et la séquence naturelle ne soit que de 38,9% (CM) et 42,3% (IF1),Cependant, l’analyse d’intégration du modèle de langage protéique montre que sa distribution dans l’espace bidimensionnel est très cohérente avec la séquence naturelle.Cela indique que ces séquences conçues sont des membres crédibles de la famille de protéines cibles, validant les capacités de conception de l'algorithme pour les familles de protéines naturelles.
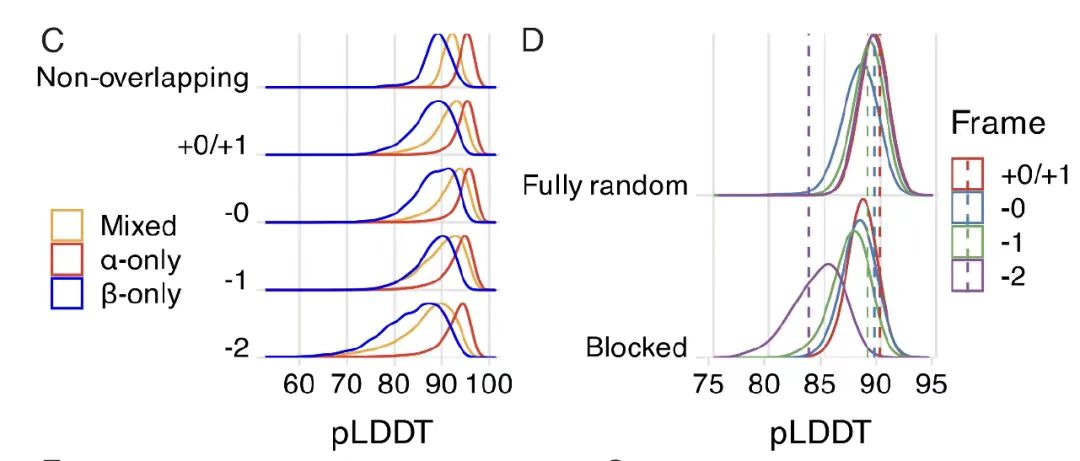
Lors de l'exploration de la faisabilité de structures protéiques hautement ordonnées et superposées,Comme le montre la figure A ci-dessous, les chercheurs ont utilisé le modèle de génération conditionnelle de structure ProteinMPNN pour générer 56 250 conceptions superposées et 33 000 conceptions non superposées pour 15 structures de chaîne principale générées de novo (couvrant les catégories de pliage α, β et mixtes). Comme le montre la figure B ci-dessous, les données d’évaluation d’AlphaFold2 montrent queLa valeur moyenne de pLDDT pour la conception avec chevauchement était de 90,2, ce qui était proche de 92,0 pour la conception sans chevauchement.
Une analyse plus approfondie a révélé que, comme le montre la figure CD ci-dessous, seule la trame -2 a eu de mauvaises performances en raison de la faible efficacité de la dégénérescence des codons. L'analyse du code génétique randomisé a montré que le code génétique naturel (SGC) présente un avantage significatif dans le codage de l'OLG, fonctionne bien sauf pour le cadre -2 et présente une préférence de composition pour les acides aminés hautement dégénérés.Le mécanisme par lequel la structure du SGC affecte la faisabilité des séquences qui se chevauchent a été révélé.
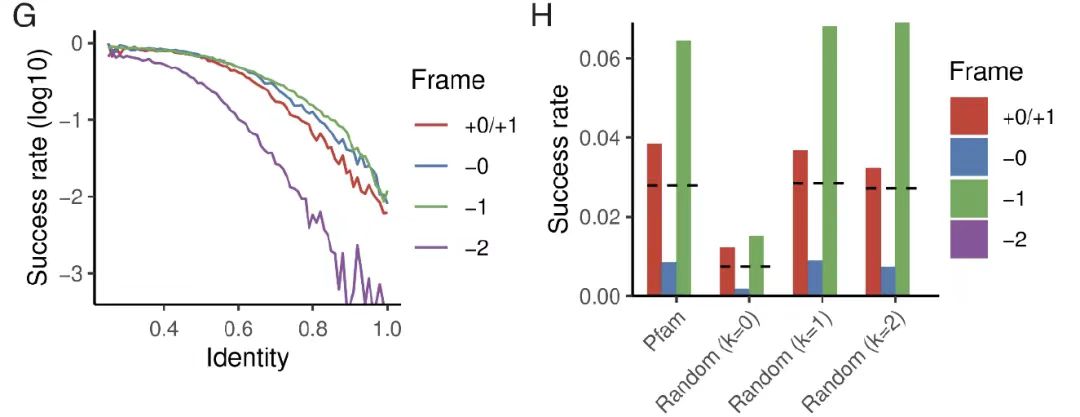
Dans les études d’accessibilité évolutive,L’équipe de recherche a commencé avec une séquence de protéines de semence avec un nombre fixe de mutations. Comme le montre la figure GF suivante,L'étude a révélé que même dans des conditions extrêmes de mutation zéro, environ 11 modèles TP3T peuvent encore atteindre une stabilité structurelle élevée (pLDDT>85, TM>0,7) ;Lorsque des séquences Pfam naturelles ont été utilisées comme parents, le taux de réussite a augmenté jusqu'à 3%, et ce résultat était cohérent avec les séquences aléatoires qui conservaient l'écart de composition du premier ordre. Cela démontre clairement que les protéines naturelles hautement optimisées peuvent accueillir de nouvelles protéines dans des cadres alternatifs sans changements de séquence majeurs, vérifiant la faisabilité de l'OLG au niveau évolutif.
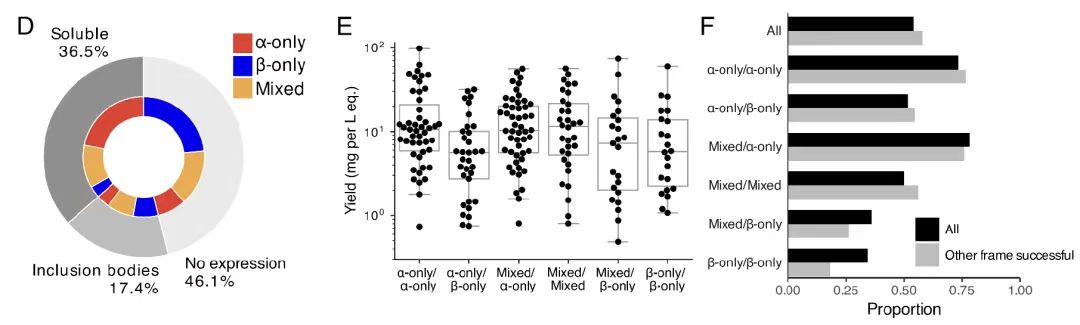
Dans la partie finale de vérification expérimentale, l’équipe de recherche a effectué l’expression recombinante et la caractérisation structurelle de 192 séquences chevauchantes. Les résultats montrent que, comme le montre la figure B,Les protéines individuelles de 54% ont été exprimées avec succès et la plupart avaient les structures secondaires attendues et une stabilité thermique élevée.
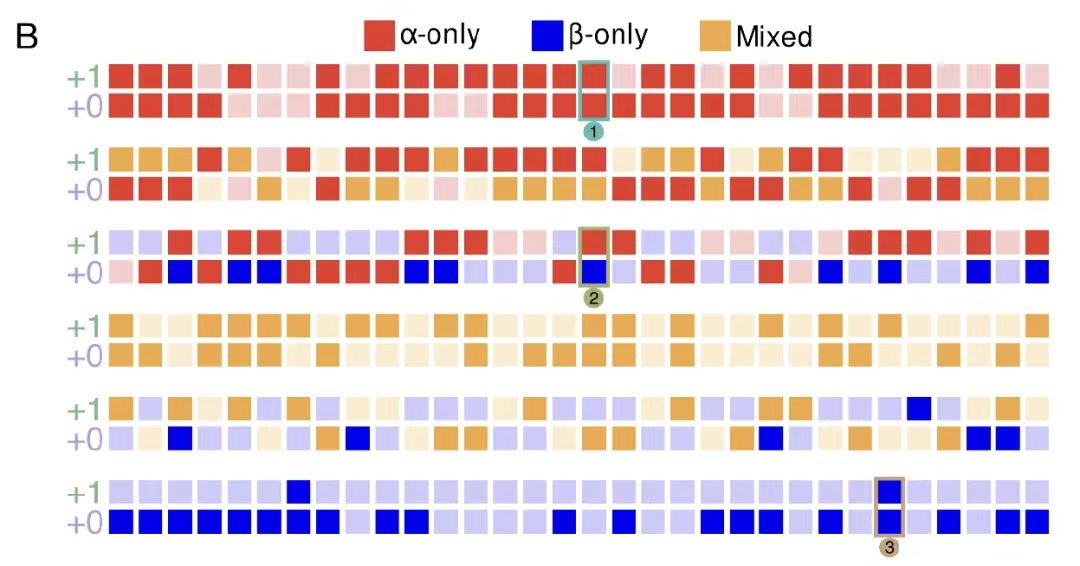
De plus, comme le montrent les figures DF ci-dessous, le taux de réussite varie en fonction du contenu de la structure secondaire de la protéine, les protéines α-hélicoïdales ayant le taux de réussite le plus élevé. De plus, les paires superposées de 31% ont été purifiées avec succès, et le succès d’une structure n’a pas affecté le succès de l’autre.Ces résultats confirment la faisabilité élevée et le taux de validation expérimentale des séquences OLG, démontrant l’efficacité de l’algorithme dans la conception de protéines chevauchantes fonctionnelles et structurellement stables.
L'exploration des frontières dans le domaine de la biologie synthétique, les applications d'ingénierie OLG s'approfondissent progressivement
Dans le domaine de la biologie synthétique, des équipes de recherche et des entreprises de nombreuses régions du monde sont engagées dans une exploration approfondie des applications d’ingénierie des gènes chevauchants (OLG).
Par exemple, le groupe de recherche de Zhu Ting de l'Université Tsinghua a réalisé des progrès significatifs dans l'étude des systèmes biologiques miroirs et a réussi à réaliser la synthèse entièrement chimique de l'ADN polymérase Pfu miroir.Cela permet non seulement d'assembler de l'ADN miroir d'une longueur de kilobases, mais aussi de développer une technologie de stockage d'informations basée sur l'ADN miroir.Cette technologie utilise la stratégie de codage des gènes miroirs pour fournir une nouvelle idée pour la superposition fonctionnelle bidirectionnelle de l'OLG. Lorsque la structure en double hélice de l'ADN miroir transporte à la fois des informations génétiques naturelles et miroir, l'utilisation de l'espace de séquence est considérablement améliorée, fournissant une base importante pour la conception compacte de génomes artificiels.
* Lien vers l'article :https://www.nature.com/articles/s41587-021-00969-6
De plus, l’équipe de Christopher Voigt au Massachusetts Institute of Technology a développé une plateforme de biologie synthétique basée sur la conception de circuits génétiques. Ils ont réussi à assembler des voies métaboliques de manière modulaire en reconstruisant la logique de régulation des groupes de gènes procaryotes. Ce chemin technique est étroitement lié à la philosophie de conception d’OLG.Lorsque plusieurs gènes fonctionnels forment un module génétique compact par le biais de séquences qui se chevauchent, cela peut non seulement réduire la redondance du génome, mais également améliorer la stabilité du système grâce à une expression coordonnée.Par exemple, le groupe de gènes artificiels fixateurs d’azote conçu par l’équipe a adopté la stratégie OLG pour compresser les séquences codantes de plusieurs enzymes clés dans la même région d’ADN, réduisant ainsi considérablement la charge métabolique des cellules hôtes tout en garantissant l’efficacité catalytique.
* Lien vers l'article :https://www.nature.com/articles/s41467-022-33272-2
Il convient de noter que ces études révèlent non seulement l’existence généralisée de l’OLG dans l’évolution naturelle, mais vérifient également sa faisabilité biophysique par des moyens d’ingénierie. Dans l'étude présentée dans cet article, l'équipe de David Baker a utilisé un modèle d'apprentissage profond pour concevoir des OLG synthétiques, qui ont montré une stabilité structurelle comparable à celle des séquences naturelles dans les simulations informatiques. Le taux de réussite élevé de la vérification expérimentale démontre une fois de plus la compatibilité biologique du codage superposé. Cette boucle fermée complète, de la recherche fondamentale à la transformation appliquée, remodèle la logique de conception de la biologie synthétique et devrait apporter de nouvelles avancées dans de nombreux domaines tels que le développement de médicaments innovants, le diagnostic de précision et la thérapie cellulaire.
Références :
1.https://www.tsinghua.edu.cn/info/1181/86148.htm
2.https://tech.huanqiu.com/article/9CaKrnJUV0x
3.https://news.bioon.com/article/4161e88572ad.html








