Command Palette
Search for a command to run...
Sélectionné Pour l'ICLR 2025, Le MIT/UC Berkeley/Harvard/Stanford Et d'autres Ont Proposé l'algorithme DRAKES Pour Briser Le Goulot d'étranglement De La Conception De Séquences Biologiques
Depuis longtemps, le principal obstacle dans le domaine de la conception des protéines n'a pas été surmonté : l'espace combinatoire des séquences d'acides aminés croît de manière exponentielle, et les méthodes de calcul traditionnelles perdent souvent de vue l'un d'eux tout en optimisant le caractère naturel et la stabilité de la séquence. Dans le domaine de la thérapie génique, les scientifiques sont également confrontés au défi de concevoir des éléments d’ADN qui régulent efficacement l’expression des gènes ; dans le développement de vaccins à ARNm, la contradiction entre l’optimisation de la séquence et l’amélioration de l’efficacité de la traduction existe toujours ; même dans les tâches de génération de langage naturel, les ingénieurs doivent rechercher un équilibre entre l'exactitude grammaticale et la sécurité du contenu. Ces défis apparemment dispersés pointent en réalité vers le même goulot d’étranglement technique :Comment optimiser des objectifs de tâches spécifiques tout en générant des séquences discrètes conformes aux distributions statistiques ?
Pour relever ce défi majeur, des chercheurs du Massachusetts Institute of Technology, de l’Université Harvard, de l’Université Stanford, de l’Université de Californie à Berkeley et de la société américaine de technologie de génie génétique Genentech ont proposé conjointement un algorithme innovant DRAKES.En introduisant un cadre d'apprentissage par renforcement, l'algorithme réalise pour la première fois la rétropropagation de récompense différentiable pour la trajectoire complète générée dans un modèle de diffusion discrète.Les expériences montrent que DRAKES peut améliorer considérablement les performances des tâches en aval tout en conservant le caractère naturel de la séquence. Son analyse théorique révèle en outre le chemin de solution optimal pour cette méthode en équilibrant la fidélité de la distribution et l'optimisation des tâches.
Les résultats de recherche associés ont été sélectionnés pour l'ICLR 2025 sous le titre « Affinage des modèles de diffusion discrète via l'optimisation des récompenses avec des applications à la conception d'ADN et de protéines ». Depuis longtemps, le principal obstacle dans le domaine de la conception des protéines n'a pas été surmonté : l'espace combinatoire des séquences d'acides aminés croît de manière exponentielle, et les méthodes de calcul traditionnelles perdent souvent de vue l'un d'eux tout en optimisant le caractère naturel et la stabilité de la séquence. Dans le domaine de la thérapie génique, les scientifiques sont également confrontés au défi de concevoir des éléments d’ADN qui régulent efficacement l’expression des gènes ; dans le développement de vaccins à ARNm, la contradiction entre l’optimisation de la séquence et l’amélioration de l’efficacité de la traduction existe toujours ; même dans les tâches de génération de langage naturel, les ingénieurs doivent rechercher un équilibre entre l'exactitude grammaticale et la sécurité du contenu. Ces défis apparemment dispersés pointent en réalité vers le même goulot d’étranglement technique :Comment optimiser des objectifs de tâches spécifiques tout en générant des séquences discrètes conformes aux distributions statistiques ?
Pour relever ce défi majeur, des chercheurs du Massachusetts Institute of Technology, de l’Université Harvard, de l’Université Stanford, de l’Université de Californie à Berkeley et de la société américaine de technologie de génie génétique Genentech ont proposé conjointement un algorithme innovant DRAKES.En introduisant un cadre d'apprentissage par renforcement, l'algorithme réalise pour la première fois la rétropropagation de récompense différentiable pour la trajectoire complète générée dans un modèle de diffusion discrète.Les expériences montrent que DRAKES peut améliorer considérablement les performances des tâches en aval tout en conservant le caractère naturel de la séquence. Son analyse théorique révèle en outre le chemin de solution optimal pour cette méthode en équilibrant la fidélité de la distribution et l'optimisation des tâches.
Les résultats de recherche associés ont été sélectionnés pour l'ICLR 2025 sous le titre « Affinage des modèles de diffusion discrète via l'optimisation des récompenses avec des applications à la conception d'ADN et de protéines ».
Adresse du document :
https://doi.org/10.48550/arXiv.2410.13643
Suivez le compte public « HyperAI Super Neural » et répondez « DRAKES » pour obtenir le PDF complet
Projet open source « awesome-ai4s »Il rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
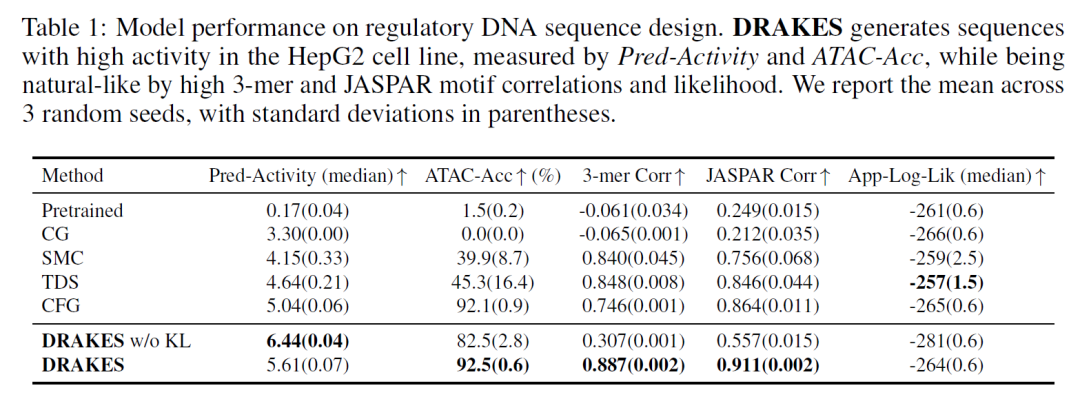
Ensemble de données : plusieurs ensembles de données sont utilisés en combinaison pour réaliser une évaluation multidimensionnelle des performances de DRAKES
Cette recherche s'est concentrée sur la séquence d'ADN régulatrice et la conception de séquences protéiques, en utilisant plusieurs ensembles de données publiques pour soutenir la validation expérimentale. Dans la conception des séquences d’ADN régulatrices, l’étude a utilisé un ensemble de données d’amplificateurs à grande échelle, qui contient environ 700 000 séquences d’ADN de 200 pb de longueur. Grâce à des essais de rapporteurs massivement parallèles (MPRA), l'activité de l'amplificateur dans les lignées cellulaires humaines a été mesurée, fournissant des données de base pour la pré-formation du modèle et la construction d'un oracle de récompense.
L'expérience a également introduit les données d'accessibilité à la chromatine de la lignée cellulaire HepG2.Utilisé pour évaluer indépendamment l'accessibilité de la chromatine des séquences synthétiques afin de valider la fiabilité de l'activité prédite. De plus, le profil de liaison du facteur de transcription JASPAR a été utilisé pour analyser les séquences générées à la recherche de motifs potentiels de liaison au facteur de transcription, aidant ainsi à l'analyse des caractéristiques clés de l'activité de l'amplificateur.
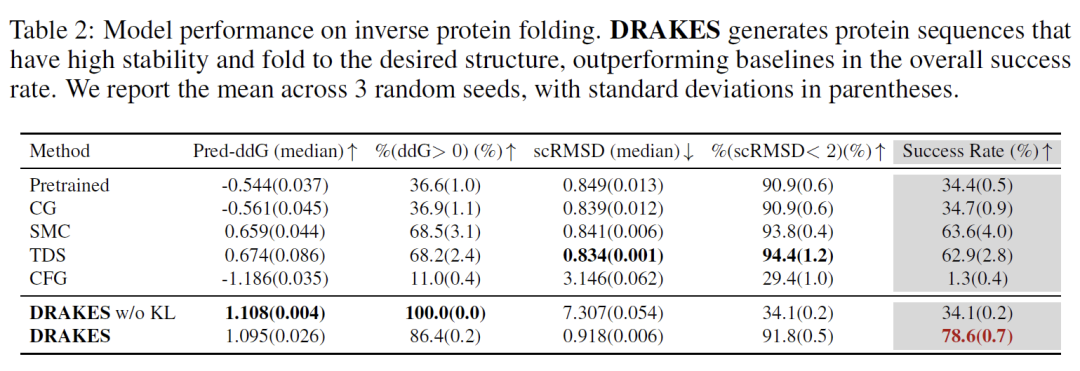
Dans la tâche de conception de séquences protéiques, le modèle de pliage inverse pré-entraîné est basé sur l'ensemble d'entraînement PDB, qui couvre la structure et les données de séquence des protéines naturelles. La formation de l'oracle de récompense s'appuie sur l'ensemble de données Megascale.L'ensemble de données contient environ 1,8 million de variantes de séquences provenant de 983 domaines naturels et conçus.Des mesures de stabilité sont fournies pour évaluer les propriétés fonctionnelles des séquences générées. Après que les données ont été examinées et divisées à l’aide de processus standard, environ 500 000 séquences de 333 domaines ont été formées, qui ont été utilisées pour construire un modèle de récompense pour le réglage fin et l’évaluation. L'utilisation combinée de ces ensembles de données garantit que la recherche peut vérifier efficacement la fonctionnalité, la similitude naturelle et la stabilité des séquences générées par le modèle dans différentes tâches de conception de biomolécules, fournissant un support empirique multidimensionnel pour l'évaluation des performances de la méthode DRAKES.
Algorithme DRAKES : adopte une architecture en deux étapes et des expériences doubles pour vérifier son potentiel d'application dans des scénarios biomédicaux
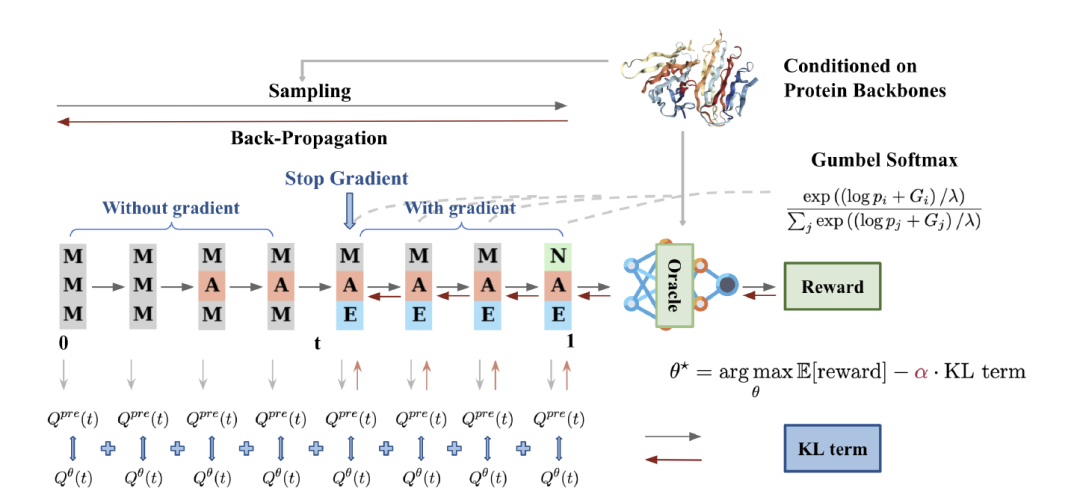
Les chercheurs ont proposé un algorithme appelé DRAKES pour affiner les modèles de diffusion discrète afin d'optimiser la fonction de récompense pour des objectifs de tâches spécifiques.L'algorithme combine le cadre d'apprentissage par renforcement (RL) et Gumbel-Softmax.L’équilibre entre la maximisation de la récompense et le maintien du caractère naturel dans les modèles de diffusion discrète est résolu. L'idée principale de DRAKES est de garantir que la séquence générée reste similaire à la distribution du modèle pré-entraîné tout en optimisant la récompense en introduisant la contrainte de divergence KL.
Plus précisément, DRAKES adopte une architecture en deux étapes, conçue respectivement pour le processus d'échantillonnage et le processus d'optimisation. Dans la phase d'échantillonnage des données, l'algorithme génère des trajectoires à travers une chaîne de Markov à temps continu (CTMC) et utilise la technique Gumbel-Softmax pour transformer le processus d'échantillonnage discret en opérations différentiables. Cette technique approxime la distribution de classification via softmax, en maintenant l'authenticité de l'échantillonnage et en conservant les informations de gradient aux paramètres de basse température.Cette conception brise la limitation de la non-différenciabilité dans les modèles de diffusion discrets traditionnels.Il fournit une base théorique pour l’optimisation ultérieure.
Dans la phase d'optimisation,L'algorithme met à jour les paramètres en maximisant la fonction objective empirique.La combinaison de la rétropropagation tronquée et de la technologie Gumbel Softmax directe peut améliorer efficacement l'efficacité de la formation. Cette architecture assure non seulement le caractère naturel des séquences générées, mais évite également le risque de sur-optimisation grâce à la contrainte de divergence KL, réalisant ainsi un équilibre dynamique entre la maximisation de la récompense et la fidélité de la distribution.
Pour vérifier l’efficacité de l’algorithme DRAKES, les chercheurs ont mené une évaluation expérimentale complète dans deux tâches clés : la conception de séquences d’ADN régulatrices et la conception de séquences protéiques.Les résultats expérimentaux démontrent systématiquement la capacité de DRAKES à optimiser de manière significative les propriétés cibles tout en préservant le caractère naturel de la séquence.
Dans la tâche d'optimisation de la séquence d'ADN régulatrice, les séquences amplificatrices générées par DRAKES ont montré des améliorations synergiques de l'activité prédite (Pred-Activity = 0,78) et de l'accessibilité à la chromatine (ATAC-Acc = 0,81) dans la lignée cellulaire HepG2, tout en maintenant la corrélation des nucléotides triplets (0,92) et la corrélation du motif JASPAR (0,88) proche de la séquence naturelle. Il convient de noter que bien que la version sans régularisation KL ait atteint une activité prédictive plus élevée (Pred-Activity=0,85), ses performances sur l'indicateur de validation indépendant ATAC-Acc (0,72) ont diminué, révélant le risque qu'une sur-optimisation puisse entraîner une déviation des séquences générées par rapport à la distribution naturelle.
Dans la tâche d'optimisation de la stabilité des protéines, les séquences générées par DRAKES ont atteint le meilleur équilibre entre la stabilité prédite (Pred-ddG = -1,23 kcal/mol) et la cohérence structurelle (taux de réussite de scRMSD < 2 83%). Des expériences comparatives montrent que bien que la version sans régularisation KL soit plus performante en termes de stabilité prédictive (Pred-ddG=-1,45 kcal/mol), son auto-cohérence structurelle est significativement réduite (le taux de réussite scRMSD<2 n'est que de 61%). Vérifié par simulation physique PyRosetta, l'énergie libre de Gibbs (ΔG=-15,2 kcal/mol) de la séquence générée par DRAKES sous la structure de chaîne principale cible est 21% inférieure à celle de la méthode de base, confirmant davantage la rationalité physique de ses résultats d'optimisation.
Les résultats expérimentaux montrent que l'algorithme DRAKES maintient le caractère naturel de la séquence (log-vraisemblance App-Log-Lik=-1,05).Les capacités d’optimisation des attributs cibles ont été considérablement améliorées.Dans la conception des éléments régulateurs des gènes, l'activité amplificatrice est améliorée par 35% ; dans la conception de médicaments protéiques, la stabilité est améliorée par 28%. Ces résultats valident non seulement le potentiel d’application de DRAKES dans des scénarios biomédicaux clés, mais établissent également un nouveau paradigme technique pour les tâches d’optimisation de séquence basées sur des modèles de diffusion discrète.
Les avancées innovantes de la Chine dans les modèles de diffusion discrète et la conception de séquences biologiques
Ces dernières années, la Chine a construit un système technique complet allant de l’innovation théorique à l’application industrielle dans le domaine des modèles de diffusion discrète et de la conception de séquences biologiques, et a proposé un certain nombre de méthodes originales dans le cadre théorique des modèles de diffusion discrète. Par exemple, le modèle de diffusion discrète hyperbolique de l'ARN tridimensionnel développé par Shanghai Yuanma Intelligent Pharmaceuticals intègre les caractéristiques géométriques de l'ARN dans l'espace hyperbolique et utilise les caractéristiques de croissance exponentielle de la géométrie hyperbolique pour obtenir une cartographie précise de la structure-séquence dans des conditions d'échantillon finies. Les données expérimentales montrent queLa similarité entre la séquence générée et la structure cible est 23% supérieure à celle de la méthode traditionnelle.Il présente des avantages significatifs, notamment dans la prédiction de structures de pseudo-nœuds complexes.Cette approche innovante d’intégration de la géométrie différentielle aux modèles génératifs marque l’entrée de la Chine dans une nouvelle étape de « paradigme auto-défini » dans le domaine de l’informatique biomoléculaire.
Dans le domaine de la thérapie génique,Le médicament pour le traitement de la surdité héréditaire développé par l'équipe de Li Huawei à l'Université Fudan.En régulant précisément l’expression fonctionnelle des séquences d’ADN, un taux d’amélioration de l’audition de 68% a été atteint lors d’essais cliniques.Le cœur de sa technologie réside dans l'établissement d'un système d'optimisation à trois niveaux « édition de séquences-régulation épigénétique-vérification fonctionnelle ».Il s'intègre parfaitement au concept d'optimisation directionnelle du modèle de diffusion discrète au niveau méthodologique. Cette avancée est due au soutien politique des « Mesures de soutien à l'industrie pharmaceutique et de la santé du groupe Changping de la zone pilote de libre-échange de Chine (Pékin) » (2023), qui répertorient clairement la thérapie cellulaire et génique comme une orientation clé et exigent une innovation collaborative complète de la « conception d'algorithmes-vérification expérimentale-transformation clinique ».
Lien vers l'article :
https://doi.org/10.1016/S0140-6736(23)02874-X
La plate-forme informatique dédiée déployée par le Centre national chinois de bioinformatique (CNCB) fournit une infrastructure stratégique pour la conception de séquences biologiques à grande échelle et peut rapidement réaliser des simulations de repliement des protéines qui prendraient des mois dans les laboratoires traditionnels. La première phase de recherche du Consortium pan-génome chinois (CPC), publiée conjointement par 26 institutions, dont l'Université Fudan, l'Université Jiaotong de Xi'an et l'Académie chinoise des sciences médicales, a initialement construit la première carte de référence pan-génome exclusive à la population chinoise, jetant les bases du déchiffrement du code génétique de la population chinoise.Ce modèle à double entraînement « puissance de calcul + données » résout efficacement les deux principaux problèmes de la conception de séquences biologiques : le problème de la spécificité de la population et la percée de l'effet longue traîne.
Face aux risques potentiels des séquences biologiques générées par l'IA, l'Assemblée populaire nationale a révisé la « Loi sur la biosécurité de la République populaire de Chine » en 2024, en mettant l'accent sur « la prévention des risques de biosécurité causés par l'abus de la technologie de l'intelligence artificielle ».Il est nécessaire de mettre en œuvre une supervision complète de la chaîne de technologies telles que l’édition génétique et la biologie synthétique.Fixer des limites sûres pour le développement technologique.
À l’heure actuelle, la Chine a formé une chaîne d’innovation complète de « théorie-application-installations-normes » dans les domaines des modèles de diffusion discrète et de la conception de séquences biologiques. Ces avancées vont non seulement remodeler la logique sous-jacente de la recherche et du développement biomédicaux, mais sont également susceptibles de donner naissance à une nouvelle génération de révolution dans l’industrie biotechnologique. Comme l'a indiqué le journal saoudien Mecca : « La Chine ne se contente pas de rattraper l'Occident, elle se dote également de ses propres capacités d'innovation. La jeune génération d'innovateurs se concentre sur les technologies de pointe, ce qui fait de la Chine un leader mondial des biotechnologies et devrait devenir une puissance mondiale. »
Références :
1.https://export.shobserver.com/baijiahao/html/709277.html
2.https://www.ncsti.gov.cn/kjdt/yqdy/cpy2/zchj/202410/t20241012_181850.html
3.https://sghexport.shobserver.com/html/baijiahao/2023/06/15/1051928.html
4.http://news.china.com.cn/2025-01/03/content_117643069.shtml