Command Palette
Search for a command to run...
L'équipe De l'Université De Tokyo a Développé Un Cadre d'apprentissage Profond STAIG Pour Éliminer Les Effets De Lot Sans Pré-alignement, Révélant Des Informations Génétiques Détaillées Dans Le Microenvironnement Tumoral

Les tissus biologiques sont des réseaux complexes composés de plusieurs types de cellules qui remplissent des fonctions importantes grâce à des configurations spatiales spécifiques. Ces dernières années, les progrès des technologies de transcriptomique spatiale (ST) telles que 10x Visium, Slide-seq, Stereo-seq et STARmap ont permis aux biologistes de cartographier les données génétiques au sein de structures spatiales, offrant ainsi des informations plus approfondies sur diverses maladies.
Cependant, les techniques ST s’appuient largement sur l’identification de régions spatiales présentant une expression génétique et des caractéristiques histologiques uniformes. à l'heure actuelle,Il existe deux principales méthodes d’identification : les méthodes de clustering non spatial et les méthodes de clustering spatial.Les méthodes de clustering non spatiales effectuent uniquement un clustering basé sur l’expression des gènes, ce qui conduit souvent à des résultats de clustering incohérents ; les méthodes de regroupement spatial utilisent des modèles de convolution de graphes pour intégrer les informations génétiques et spatiales, mais s'appuient sur des normes de distance définies artificiellement lors de la conversion des données ST en structures de graphes, ce qui peut introduire un biais ; dans le même temps, les méthodes utilisant des images histologiques sont également confrontées à des défis car elles sont sensibles aux changements de qualité de coloration ; en outre, l'intégration par lots de la plupart des méthodes existantes nécessite encore une intervention manuelle, comme l'alignement manuel des coordonnées ou le recours à des outils supplémentaires.
Pour surmonter ces défis,Une équipe de recherche de l'Institut des sciences médicales de l'Université de Tokyo, au Japon, a proposé un cadre d'apprentissage en profondeur appelé STAIG (Spatial Transcriptomics Analysis with Image-Assisted Graph Comparative Learning).Capacité à intégrer l'expression génétique, les données spatiales et les images histologiques sans alignement.
STAIG extrait des caractéristiques d'images colorées à l'hématoxyline et à l'éosine (H&E) via un modèle auto-supervisé sans s'appuyer sur des ensembles de données histologiques à grande échelle pour la pré-formation. De plus, STAIG ajuste dynamiquement la structure du graphique pendant la formation et utilise les informations de l'image histologique pour exclure sélectivement les échantillons négatifs homologues, réduisant ainsi le biais introduit par la construction initiale.
Enfin, STAIG identifie les points communs dans l'expression des gènes grâce à une comparaison locale, permettant une intégration de lots de bout en bout sans nécessiter d'alignement manuel des coordonnées et réduisant efficacement les effets de lots. Les chercheurs ont évalué STAIG sur plusieurs ensembles de données.Les résultats ont montré qu'il présente de bonnes performances dans l'identification des régions spatiales et peut révéler des informations spatiales et génétiques détaillées dans le microenvironnement tumoral, favorisant ainsi la compréhension de systèmes biologiques complexes.
Les résultats associés ont été publiés dans Nature Communications sous le titre « STAIG : Analyse transcriptomique spatiale via l'apprentissage contrastif de graphes assisté par image pour l'exploration de domaine et l'intégration sans alignement ».
Points saillants de la recherche :
* Le modèle STAIG permet l'intégration de sections de tissus sans pré-alignement et élimine les effets de lot
* Le modèle STAIG est applicable aux données acquises à partir de différentes plateformes, que des images histologiques soient incluses ou non
* Les chercheurs démontrent que STAIG peut identifier des régions spatiales avec une grande précision et révéler de nouvelles perspectives sur le microenvironnement tumoral, démontrant ainsi son large potentiel pour décortiquer la complexité biologique spatiale

Adresse du document :
https://www.nature.com/articles/s41467-025-56276-0
Adresse de téléchargement de l'ensemble de données utilisé dans cette étude :
https://go.hyper.ai/m5YC4
Ensemble de données : Collection d'ensembles de données ST et d'images histologiques provenant de différentes plateformes
Les chercheurs ont téléchargé des ensembles de données ST et des images histologiques accessibles au public à partir de différentes plateformes.Comme le montre la figure ci-dessous. L'ensemble de données ST comprend l'ensemble de données du cortex préfrontal dorsolatéral humain (DLPFC), l'ensemble de données sur le cancer du sein humain, l'ensemble de données sur le cerveau de souris, l'ensemble de données Slide-seqV2, l'ensemble de données STARmap, etc.
Adresse de téléchargement du jeu de données :
https://go.hyper.ai/m5YC4
* L'ensemble de données du cortex préfrontal dorsolatéral humain (DLPFC) de la plateforme 10x Visium contient 12 tranches de 3 individus, chacune fournissant 4 tranches avec des intervalles de 10 μm et 300 μm, et le nombre de points de chaque tranche varie de 3 498 à 4 789. Ces tranches ont été annotées manuellement comme couches corticales L1–L6 et matière blanche (WM) ;
* L’ensemble de données sur le cancer du sein humain contient 3 798 points ;
* L'ensemble de données du cerveau de la souris comprend deux tranches, antérieure et postérieure, contenant respectivement 2 695 et 3 355 points ;
* Pour le mélanome du poisson zèbre, les chercheurs ont analysé les sections A et B, qui contenaient respectivement 2 179 et 2 677 taches ;
* Pour l'expérience d'ensemble, des ensembles de données DLPFC et de cerveau de souris ont été utilisés. L'ensemble de données Stereo-seq du bulbe olfactif de la souris contient 19 109 points avec une résolution de 14 μm ;
* Ensemble de données Slide-seqV2 avec une résolution de 10 μm, incluant l'hippocampe du rat (18 765 points du quart central du rayon) et le bulbe olfactif du rat (19 285 points) ;
* L'ensemble de données STARmap contient 1 207 points ;
* Pour l'ensemble de données MERFISH, le MTG humain contient 3 970 spots, tandis que les régions VIS de la souris 1 et de la souris 2 contiennent respectivement 5 995 et 2 479 spots.
Architecture du modèle : Apprentissage du contraste des graphes assisté par image pour l'analyse transcriptomique spatiale
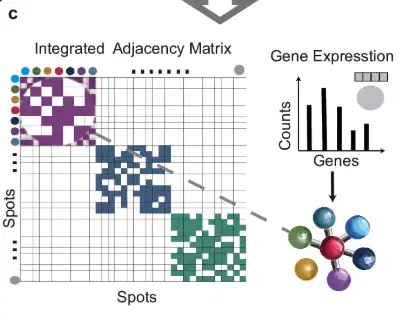
La figure ci-dessous décrit le cadre global de STAIG, un cadre d'apprentissage profond qui utilise l'apprentissage contrastif graphique combiné à l'extraction de fonctionnalités hautes performances pour intégrer l'expression génétique, les coordonnées spatiales et les images histologiques. Il se compose de 6 modules :
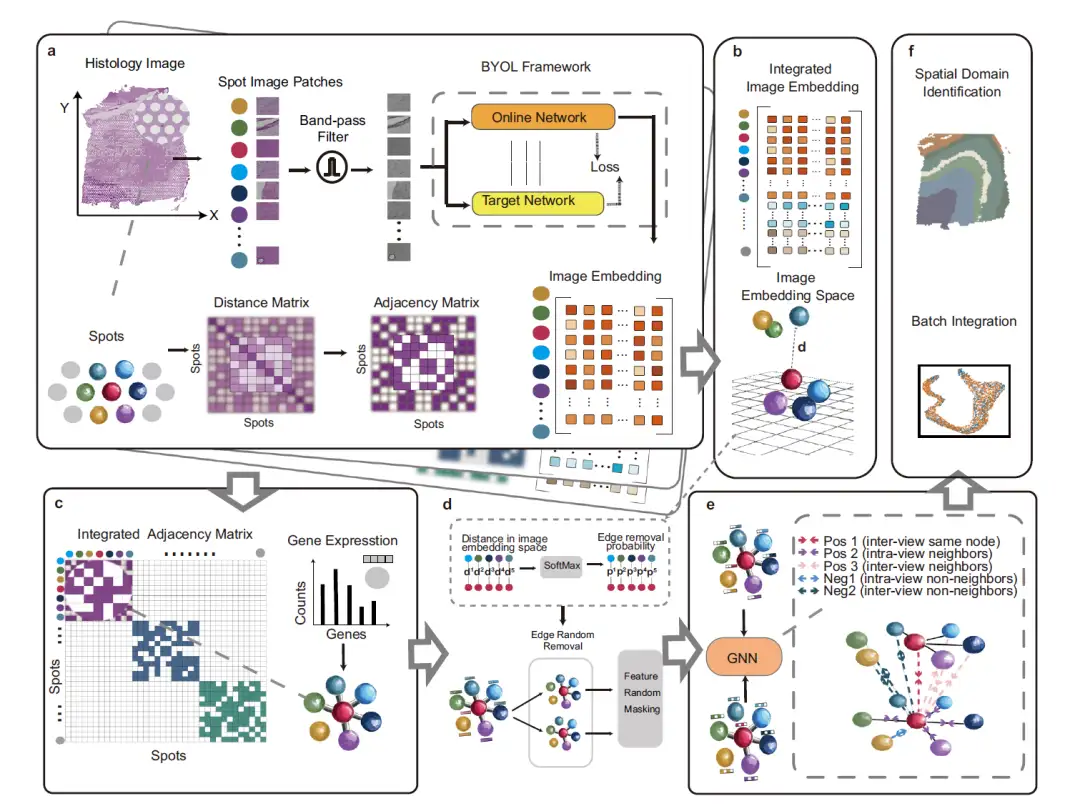
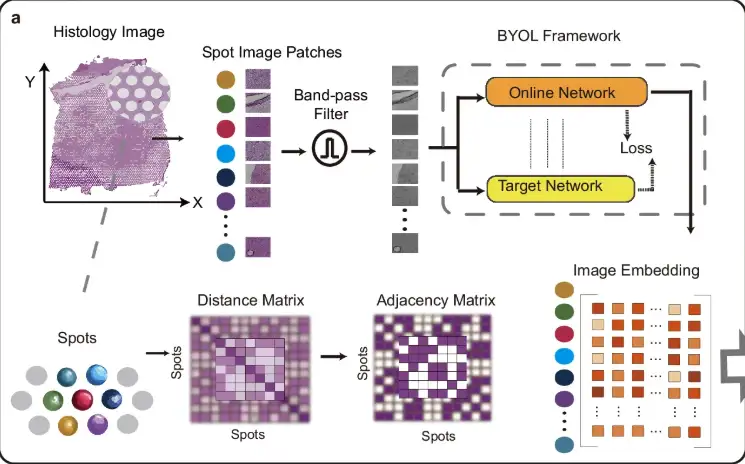
Tout d’abord, comme le montre la partie a de la figure suivante, afin de réduire l’impact du bruit et de la coloration inégale des tissus, STAIG divise d’abord l’image histologique en petits patchs (Spot Image Patches) alignés avec les positions spatiales des points de données, puis utilise un filtre passe-bande pour optimiser l’image. Les caractéristiques d'intégration d'image sont extraites via le modèle auto-supervisé Bootstrap Your Own Latent (BYOL), et une matrice d'adjacence est construite en fonction de la distance spatiale entre les points de données.
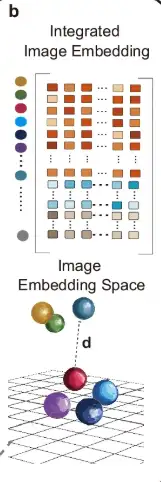
Comme le montre la partie b de la figure suivante, afin d’intégrer les données de différentes sections de tissus, STAIG utilise une méthode d’empilement vertical pour intégrer les caractéristiques de plusieurs sections de tissus.
Comme le montre la partie c de la figure ci-dessous, les matrices d'adjacence de chaque tranche sont fusionnées à l'aide de la méthode de placement diagonal pour former une matrice d'adjacence intégrée, qui est ensuite utilisée pour construire une structure graphique avec des données d'expression génétique comme informations de nœud.
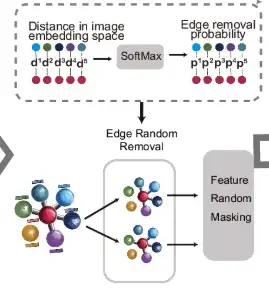
Comme le montre la partie d de la figure ci-dessous, pour les points de mesure reliés par des arêtes, leurs distances sont calculées dans l'espace d'intégration de l'image, et la fonction SoftMax est utilisée pour convertir ces distances en probabilité de suppression aléatoire des arêtes. Sur cette base, le graphique d'origine subit deux cycles de suppression aléatoire des bords (Edge Random Removal) pour générer deux vues améliorées. Ensuite, les fonctionnalités des nœuds dans ces vues sont masquées de manière aléatoire.
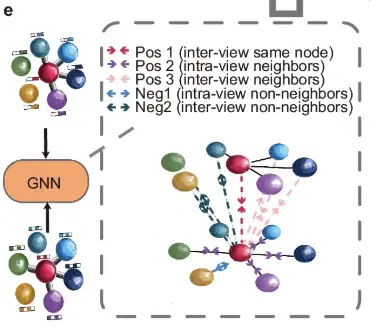
Ensuite, comme le montre la partie e de la figure, la vue augmentée est ensuite traitée par un réseau neuronal graphique partagé (GNN) et guidée par un objectif contrastif voisin, qui vise à rapprocher les nœuds voisins tout en éloignant les nœuds non voisins dans les deux vues graphiques.
Enfin, comme le montre la figure f, le GNN formé génère des intégrations pour identifier les régions spatiales et minimiser les effets de lot entre les sections de tissus consécutives.

Résultats de recherche : STAIG montre des performances supérieures dans diverses conditions
L'équipe de recherche a mené une évaluation comparative approfondie pour comparer STAIG avec d'autres techniques ST de pointe.Les résultats montrent que STAIG présente des performances supérieures dans diverses conditions.
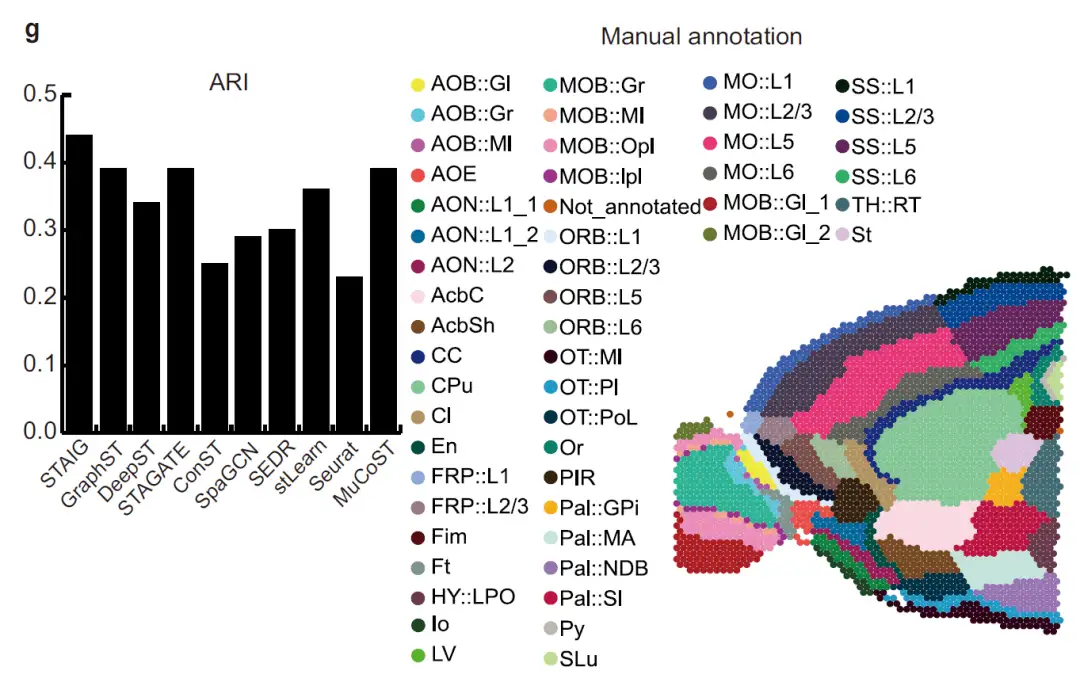
Évaluation des performances de reconnaissance des régions cérébrales
Pour évaluer les performances de STAIG dans la reconnaissance des régions tissulaires, les chercheurs ont comparé STAIG avec des méthodes existantes, notamment Seurat, GraphST, DeepST, STAGATE, SpaGCN, SEDR, ConST, MuCoST et stLearn. Les indicateurs d’évaluation de la performance comprennent :
* Indice Rand ajusté (ARI) et information mutuelle normalisée (NMI) (pour les ensembles de données annotés manuellement).
* Coefficient de silhouette (SC) et indice de Davis-Bolding (DB) (pour d'autres ensembles de données).
1 Performances de l'ensemble de données du cerveau humain
Dans l'ensemble,STAIG obtient les meilleurs résultats sur l’ensemble de données du cerveau humain.Les valeurs médianes les plus élevées de l'ARI (0,69) et du NMI (0,71) ont été atteintes, comme le montre la figure suivante :
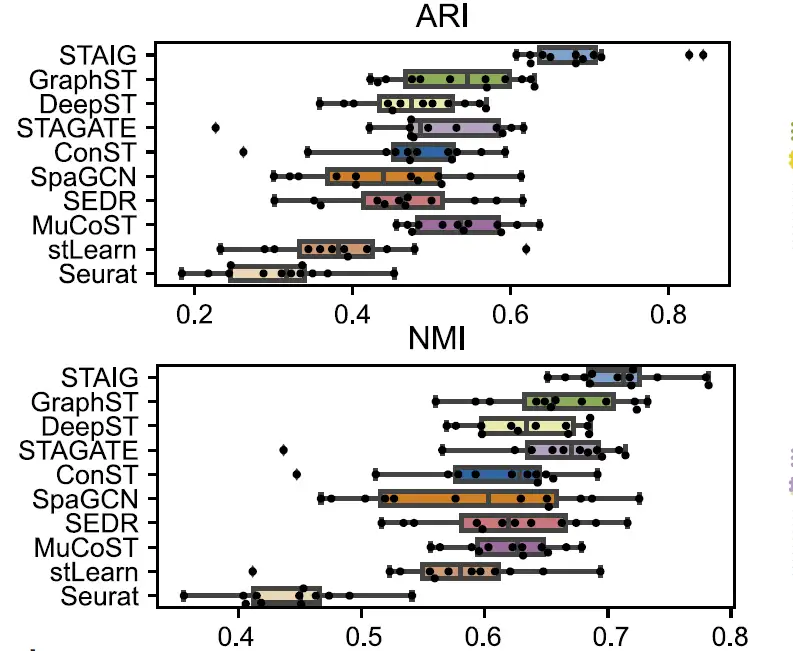
En comparaison, les méthodes existantes sont peu performantes : stLearn évalue mal certains points et manque certaines couches ; GraphST a un ARI de 0,64 et un NMI de 0,73, mais présente de grands écarts dans les positions des couches L4 et L5 ; les ARI des autres méthodes varient de 0,25 à 0,57 et les NMI varient de 0,42 à 0,69, principalement en raison d'une identification inexacte du rapport de couche.
2 Performances de l'ensemble de données du cerveau de souris
Comme le montre la figure ci-dessous, dans l’ensemble de données du cerveau postérieur de la souris,STAIG a identifié avec succès le cortex cérébelleux et l'hippocampe, et a également distingué la corne d'Ammon (CA) et le gyrus denté.Très cohérent avec l’annotation de l’atlas cérébral de la souris Allen ; malgré l'absence d'annotation manuelle, STAIG a quand même obtenu le SC le plus élevé (0,31) et le DB le plus bas (1,11), indiquant ses performances de clustering supérieures.
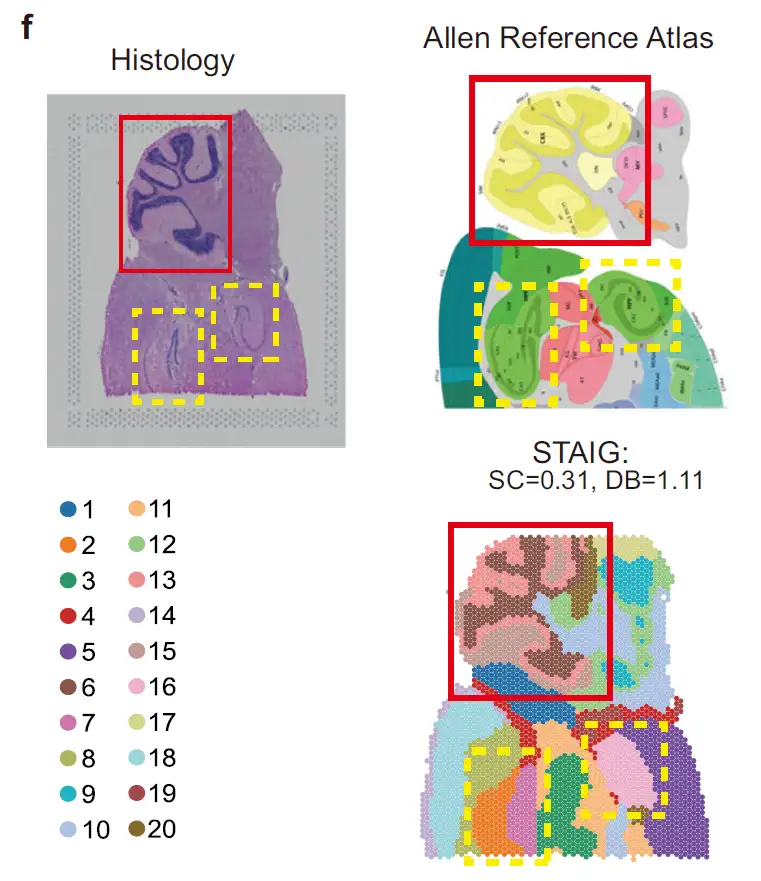
Comme le montre la figure ci-dessous, dans l’ensemble de données du cerveau antérieur de la souris,STAIG a segmenté avec précision le bulbe olfactif et le pallium dorsal.Après avoir consulté l'annotation manuelle de Long et al., son ARI a atteint 0,44 et son NMI a atteint 0,72, qui étaient tous deux les valeurs les plus élevées.
Efficacité de l'extraction des caractéristiques de l'image
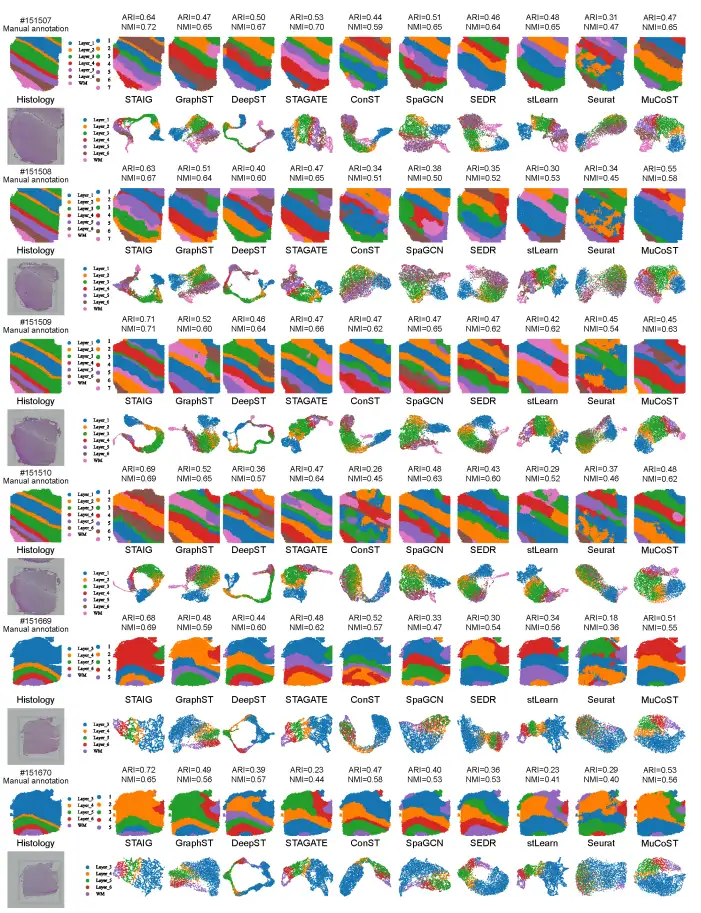
Pour explorer l’impact des caractéristiques de l’image, les chercheurs ont utilisé l’algorithme KNN pour comparer les caractéristiques de l’image extraites par STAIG avec celles extraites par d’autres méthodes (stLearn, DeepST et ConST).
1 Analyse de tranches de tissu cérébral
En prenant la tranche #151507 comme exemple, comme le montre la figure ci-dessous, les caractéristiques de l'image de stLearn sont sérieusement affectées par l'intensité de la coloration, ce qui entraîne une inadéquation avec l'annotation hiérarchique réelle ; bien que DeepST et ConST utilisent l’apprentissage profond, ils ne parviennent pas à capturer avec précision les caractéristiques de texture complexes du tissu cérébral ; les résultats d'extraction de caractéristiques de STAIG correspondent étroitement aux niveaux annotés manuellement, bien que certaines limites soient encore légèrement floues, elles ne sont presque pas affectées par les différences de coloration.

2 Analyse d'images de tissus cancéreux du sein
Les chercheurs ont également testé la capacité d’extraction des caractéristiques de l’image en utilisant des images colorées H&E de cancer du sein humain, comme le montre la figure ci-dessous.
Les résultats ont montré que l’image de stLearn présente un mélange de tumeurs et de zones normales, avec une faible discrimination ; ConST semblait diviser l'image en différentes régions, mais après un zoom avant, les limites des régions s'écartaient considérablement des annotations manuelles ; DeepST n’a pas réussi à extraire des caractéristiques d’image efficaces ;STAIG identifie avec précision les régions tumorales. Ses résultats de regroupement spatial maintiennent un degré élevé de cohérence régionale et les régions segmentées correspondent presque parfaitement aux contours annotés manuellement.A vérifié son excellente capacité d'extraction de caractéristiques d'image.

Définition du microenvironnement tumoral dans le cancer du sein humain ST
Dans une analyse d’un ensemble de données sur le cancer du sein humain,Les chercheurs ont constaté que les résultats de STAIG étaient très cohérents avec les annotations manuelles et ont atteint les ARI les plus élevés (0,64) et NMI (0,70).Il est à noter que STAIG propose une stratification spatiale légèrement différente mais plus raffinée, en particulier pour la région Healthy_1 annotée manuellement (Figure 2a), que STAIG subdivise en sous-groupes 3 et 4 (Figure 2b).
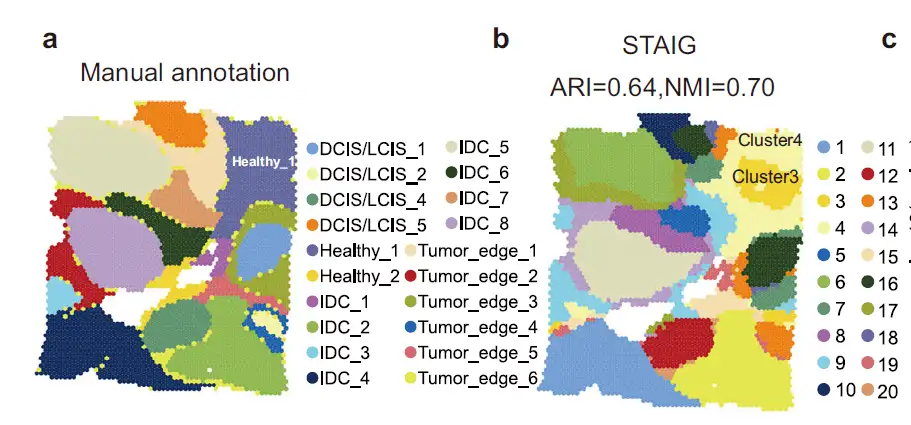
En conclusion, grâce à l’intégration multimodale de STAIG, nous avons découvert que le sous-groupe 3 formait un microenvironnement tumoral dense en CAF et révélait les caractéristiques moléculaires des zones riches en CAF.
L'apprentissage profond fournit des outils puissants pour le développement de la technologie ST
Grâce au développement rapide de la génomique et de la technologie ST, la communauté biomédicale est en mesure d’explorer la distribution spatiale de l’expression des gènes dans les tissus, révélant ainsi les fonctions et les structures complexes des organismes. La technologie ST fournit non seulement des informations quantitatives sur l’expression des gènes, mais préserve également la relation spatiale des cellules dans les tissus, permettant aux chercheurs d’étudier le microenvironnement tissulaire, les interactions cellulaires et les caractéristiques spatiales du développement de la maladie. Cependant,Étant donné que les données ST présentent généralement des problèmes tels qu'une dimensionnalité élevée, un bruit important et des effets de lot, la manière d'intégrer et d'analyser efficacement ces données est devenue un défi majeur dans la recherche actuelle.
L'introduction de techniques d'apprentissage profond, en particulier les réseaux de neurones graphiques (GNN) et les méthodes d'apprentissage contrastif, fournit des outils puissants pour l'analyse des données ST. Les méthodes d’analyse traditionnelles s’appuient souvent sur la réduction de la dimensionnalité et le clustering, tandis que les méthodes d’apprentissage en profondeur peuvent extraire automatiquement des fonctionnalités à plusieurs niveaux et optimiser la représentation des données grâce à une formation de bout en bout. Comme mentionné ci-dessus, la méthode basée sur GNN peut utiliser les informations d'adjacence spatiale pour construire une structure graphique, de sorte que le modèle peut non seulement capturer l'expression des gènes, mais également apprendre les dépendances spatiales entre les cellules. L’introduction de l’apprentissage contrastif améliore encore la capacité de généralisation du modèle, lui permettant d’apprendre des caractéristiques spatiales clés sans annotation.
En outre, l'industrie a également fait de nombreux progrès dans la combinaison de l'apprentissage profond et de la technologie ST :
Novembre 2024L'équipe dirigée par Yang Yungui du Centre national de bioinformatique de Chine et l'équipe dirigée par Zhang Shihua de l'Institut de mathématiques et de sciences des systèmes de l'Académie chinoise des sciences ont développé un outil d'annotation cellulaire du transcriptome spatial basé sur l'apprentissage en profondeur appelé STASCAN.En intégrant les profils d'expression génétique et l'apprentissage des caractéristiques cellulaires à partir d'images histologiques, nous prédisons les types de cellules dans des régions inconnues de sections de tissus et annotons les cellules dans la zone capturée, améliorant ainsi considérablement la résolution cellulaire spatiale. De plus, STASCAN est applicable à différents ensembles de données provenant de différentes techniques ST et présente des avantages significatifs dans le déchiffrement de la distribution cellulaire à haute résolution et la résolution de l'architecture tissulaire améliorée.
Ce résultat a été publié dans Genome Biology sous le titre « STASCAN déchiffre les cartes de distribution cellulaire à résolution fine en transcriptomique spatiale par apprentissage profond ».
* Adresse du papier :
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03421-5
23 janvier 2025Une équipe de recherche de l'Université de Princeton aux États-Unis a développé un nouvel algorithme d'apprentissage profond GASTON (Gradient Analysis of Spatial Transcriptomics Organization with Neural networks). En combinant des réseaux neuronaux profonds non supervisés avec des algorithmes interprétables, GASTON a proposé de manière innovante le concept d'« isoprofondeur », qui est similaire à l'altitude sur une carte topographique et est utilisé pour quantifier la structure topologique spatiale de l'expression des gènes dans les coupes de tissus. ,
Grâce à l'iso-profondeur et à son gradient, les chercheurs peuvent non seulement segmenter différentes régions spatiales du tissu, mais également identifier les tendances de changement continu et les gènes marqueurs clés de l'expression génétique au sein du tissu. L'étude a démontré l'application réussie de GASTON dans une variété d'échantillons biologiques, notamment le cerveau de souris, le bulbe olfactif de souris, le microenvironnement tumoral du cancer colorectal, etc. Les résultats montrent que GASTON peut analyser avec précision la structure des tissus, révéler la distribution spatiale et les modèles de changement des types de cellules, et découvrir de nombreux modèles d'expression génique spatiale qui sont négligés par d'autres méthodes.
Les résultats associés ont été publiés dans Nature Methods sous le titre « Cartographie de la topographie de l'expression spatiale des gènes avec un apprentissage profond interprétable ».
* Adresse du papier :
https://www.nature.com/articles/s41592-024-02503-3
De toute évidence, la combinaison de l’apprentissage profond et de la technologie ST améliore non seulement la capacité d’intégration des données et de réduction du bruit, mais favorise également l’exploitation approfondie des informations biologiques spatiales. À l’avenir, avec la croissance des ressources informatiques et l’optimisation des algorithmes, l’apprentissage en profondeur jouera un rôle plus important dans l’analyse des données ST et fournira un soutien plus fort à la médecine de précision et au traitement personnalisé.
Références :
1.https://static-content.springer.com/esm/art%3A10.1038%2Fs41467-025-56276-0/MediaObjects/41467_2025_56276_MOESM1_ESM.pdf
2.https://www.bjqykxy.com/kexueyanjiu/dongwuzhiwu/7361.html
3.https://news.qq.com/rain/a/20250128A057OQ00?suid=&media_id=
4.https://www.medsci.cn/article/show_








