Command Palette
Search for a command to run...
L'université Columbia Et l'université Stanford Collaborent ! Squidiff Permet La Simulation De Transcriptomes Multi-scénarios, Contribuant Ainsi Au Développement De La Médecine De Précision Et De La Médecine spatiale.
En biologie cellulaire, les cellules vivantes sont des systèmes dissipatifs complexes, toujours loin de l'équilibre chimique. Comprendre leur réponse collective aux stimuli externes demeure une question scientifique fondamentale que les chercheurs s'efforcent d'élucider. Cette réponse est non seulement régulée conjointement par l'hétérogénéité tissulaire interne et les signaux externes, mais elle présente aussi souvent des caractéristiques dynamiques non linéaires imprévisibles. Bien que le séquençage unicellulaire permette d'analyser sans biais la composition hétérogène des cellules, le suivi précis des modifications du transcriptome complet après stimulation reste un défi majeur.
Pour pallier cette limitation, la communauté scientifique a développé divers modèles d'apprentissage automatique, tels que scGen et CellOT. Cependant, ces modèles peinent à prédire les transitions dynamiques à haute résolution et la plupart reposent sur une conception spécifique à la tâche, ce qui limite considérablement leur applicabilité. L'avènement des modèles de diffusion a constitué une avancée majeure dans ce domaine : la génération itérative de données optimisées permet de capturer des caractéristiques de distribution de données plus riches, offrant ainsi une nouvelle approche pour résoudre les problèmes susmentionnés. Actuellement, certaines études tentent de combiner les modèles de diffusion avec des auto-encodeurs variationnels (VAE) ou d'implémenter le processus de diffusion dans l'espace latent, générant avec succès des données unicellulaires de haute fidélité et améliorant l'efficacité de la modélisation.Cependant, l'application des modèles de diffusion dans des scénarios clés tels que la prédiction de la réponse à la perturbation génétique, la prédiction de la réponse à la perturbation médicamenteuse et l'inférence de la trajectoire de développement cellulaire reste un domaine sous-développé..
Dans ce contexte,Des équipes de recherche de l'Université Columbia, de l'Université Stanford et d'autres institutions ont développé le cadre de calcul Squidiff.Ce cadre est construit sur un modèle implicite de diffusion débruité conditionnellement et peut prédire les réponses transcriptomiques de différents types de cellules sous induction de différenciation, perturbation génétique et traitement médicamenteux.Son principal avantage réside dans sa capacité à intégrer des informations définitives provenant d'outils d'édition génique et de composés médicamenteux :Pour prédire la différenciation des cellules souches, Squidiff permet non seulement de saisir avec précision les états cellulaires transitoires, mais aussi d'identifier les effets non additifs des perturbations génétiques et les caractéristiques de réponse spécifiques à chaque type cellulaire. L'équipe de recherche a ensuite appliqué Squidiff à l'étude des organoïdes vasculaires, prédisant avec succès les effets de l'exposition aux radiations sur différents types cellulaires et évaluant l'efficacité protectrice des médicaments radioprotecteurs.
Les résultats de cette recherche, intitulée « Squidiff : prédiction du développement cellulaire et des réponses aux perturbations à l'aide d'un modèle de diffusion », ont été publiés dans Nature Methods.

Adresse du document :
https://www.nature.com/articles/s41592-025-02877-y
Suivez notre compte WeChat officiel et répondez « Squidiff » en arrière-plan pour obtenir le PDF complet.
Autres articles sur les frontières de l'IA :
https://hyper.ai/papers
Ensemble de données : Couverture complète de plusieurs scénarios + contrôle qualité standardisé
Pour former et valider pleinement les performances du framework Squidiff,L'équipe de recherche a construit un ensemble de données multi-scénarios comprenant des données expérimentales simulées et réelles, couvrant des axes de recherche clés tels que la différenciation cellulaire, la perturbation génétique, le traitement médicamenteux et la réponse aux radiations des organoïdes vasculaires.Toutes les données ont fait l'objet d'un contrôle qualité uniforme : les cellules de faible qualité présentant un ratio de gènes mitochondriaux supérieur à 20% ou un nombre de gènes inférieur à 1 000 ont été éliminées, les gènes faiblement exprimés ont été supprimés et, dans certains cas, les gènes présents dans les cellules doubles et les gènes liés au stress ont été exclus. Enfin, les différences de profondeur de séquençage ont été corrigées par normalisation logarithmique afin de garantir la comparabilité des jeux de données.
Concernant les données simulées, l'équipe a utilisé l'outil Splatter basé sur une distribution gamma-Poisson hiérarchique pour générer des données synthétiques de séquençage d'ARN monocellulaire, simulant l'hétérogénéité d'expression et les caractéristiques de variance du scRNA-seq réel, afin de vérifier les capacités de base du modèle en matière de reconstruction et d'inférence du transcriptome sans avoir besoin d'un prétraitement biologique supplémentaire.
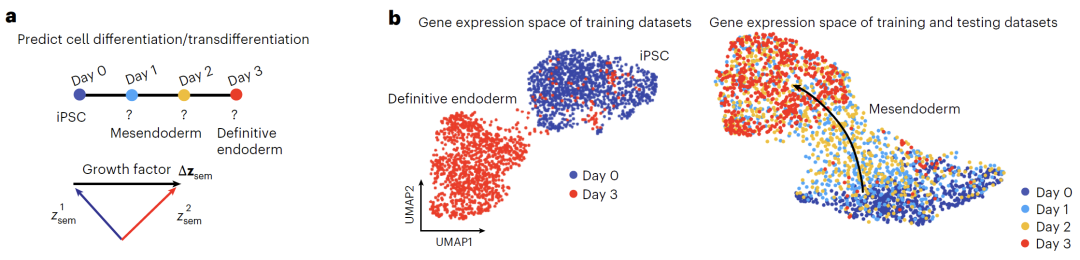
Les données de différenciation cellulaire proviennent d'un jeu de données public de différenciation de cellules souches pluripotentes induites (iPSC) humaines en endoderme, contenant les transcriptomes de 4 800 cellules du jour 0 (état iPSC) au jour 3 (état endodermique défini). Le modèle a utilisé les données des jours 0 et 3 comme ensemble d'entraînement et celles des jours 1 et 2 comme ensemble de test. Les 203 gènes les plus hypervariables ont été sélectionnés pour la modélisation. Un bruit gaussien a été introduit lors de l'entraînement, et 1 000 étapes de diffusion ont été définies. Les variables sémantiques de différenciation ont été obtenues en calculant la différence moyenne des représentations latentes, puis une interpolation linéaire a été utilisée pour simuler la trajectoire de développement du jour 0 au jour 3 afin d'évaluer la capacité prédictive du modèle pour le processus de différenciation dynamique.
Les données relatives à la perturbation génétique proviennent d'une expérience de criblage CRISPR sur des cellules K562.L'étude a porté sur environ 10 000 cellules, incluant des cellules invalidées pour les gènes ZBTB25 et PTPN12 ainsi que leurs témoins de type sauvage. Les données ont été réparties en trois groupes : « PTPN12 + témoin », « ZBTB25 + témoin » et « PTPN12 + ZBTB25 ». Les deux premiers groupes ont servi à l'entraînement du modèle, et le dernier à sa validation. Après l'entraînement, les variables spécifiques à chaque perturbation génique ont été extraites et combinées afin de simuler les modifications transcriptomiques résultant de la double invalidation génique, validant ainsi la capacité du modèle à capturer les effets non additifs.
Les données de traitement des médicaments intègrent de multiples échantillons de cellules et de médicaments.Ceci inclut les profils d'expression de glioblastomes traités par six médicaments, dont l'étoposide, et les données de réponse de mélanomes à des combinaisons de médicaments. Lors de l'entraînement, le modèle apprend des représentations de perturbation spécifiques à chaque médicament et intègre des échantillons de médicaments inconnus issus de l'ensemble de données sci-Plex3. En combinant la structure SMILES, les informations de dosage et les empreintes digitales des composés, le modèle parvient à des prédictions généralisées des effets de perturbation de médicaments inconnus.
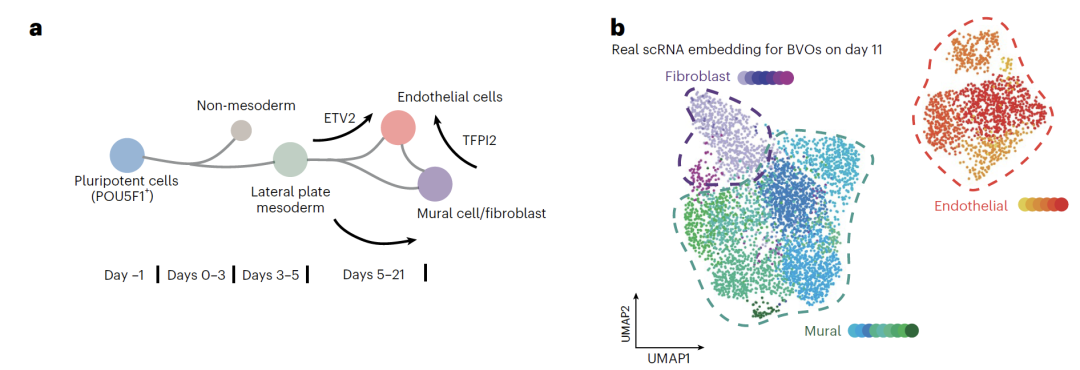
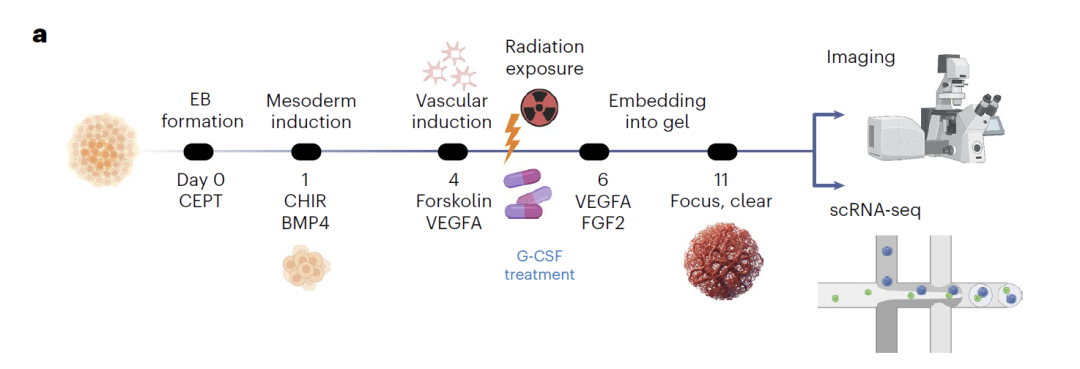
Les données relatives aux organoïdes vasculaires sont basées sur une construction expérimentale originale.Des cellules endothéliales, pariétales et des fibroblastes ont été différenciés à partir de cellules iPS humaines saines. Au jour 5, les cellules ont été soumises à une irradiation neutronique ou photonique, et des données scRNA-seq ont été collectées au jour 11, constituant une bibliothèque de ressources comprenant 72 organoïdes et environ 60 000 cellules. Une validation multimodale a été complétée par des dosages ELISA de facteurs inflammatoires. Pour la modélisation, l’équipe a utilisé les données des jours 0 et 11 afin d’entraîner le modèle et d’interpoler pour prédire l’état des cellules à des temps intermédiaires. Dans les scénarios d’irradiation et de traitement par G-CSF, seules les données des cellules endothéliales ont été utilisées pour l’entraînement, générant des transcriptomes perturbés pour les trois types cellulaires. Enfin, la signification biologique des résultats prédits a été validée par une analyse d’expression différentielle et une analyse pseudo-temporelle.
Squidiff : un modèle de diffusion conditionnelle intégrant DDIM et l’encodage sémantique
Pour prédire avec précision la réponse dynamique du transcriptome sous diverses perturbations telles que la différenciation, le développement, l'édition de gènes et le traitement médicamenteux, l'équipe de recherche a développé Squidiff, un cadre informatique intelligent basé sur le modèle de diffusion conditionnelle.Comme illustré dans la figure ci-dessous, ce modèle intègre étroitement le modèle implicite de diffusion avec débruitage conditionnel (DDIM) à une technologie d'encodage sémantique afin de construire une architecture collaborative en trois étapes : « encodage-diffusion-décodage ». Il permet non seulement de générer efficacement des données de transcriptome conformes au contexte biologique, mais aussi de réguler avec souplesse l'état cellulaire grâce à des variables latentes. Il est ainsi largement applicable à divers contextes de recherche tels que la différenciation cellulaire, la perturbation génique et les traitements médicamenteux.
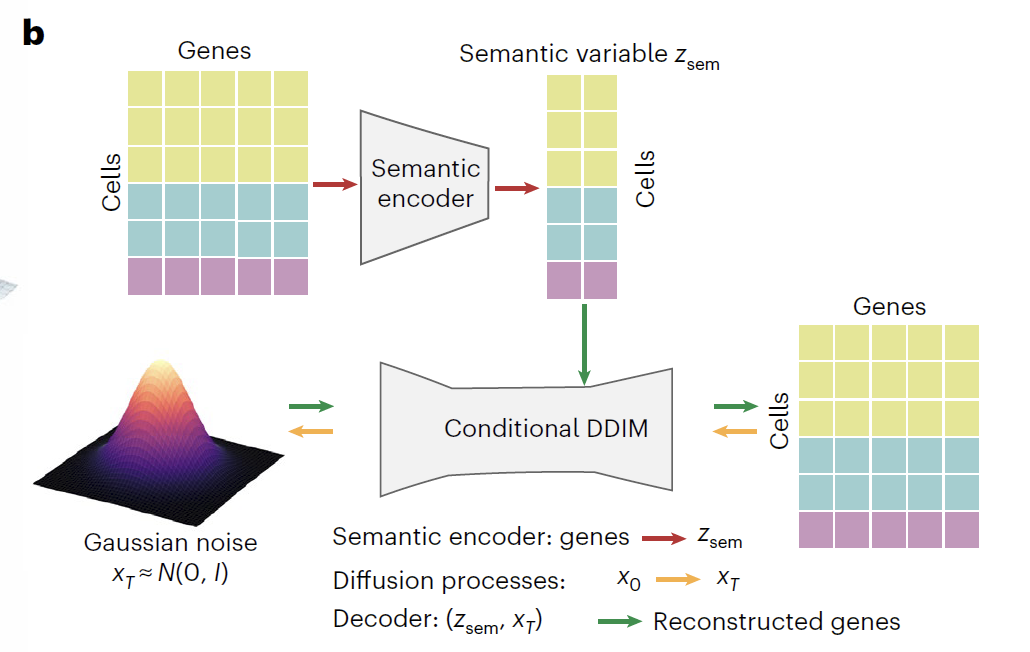
Le cœur de Squidiff repose sur un encodeur sémantique et un module de diffusion DDIM conditionnel. L'encodeur sémantique utilise un perceptron multicouche (MLP) pour projeter les données de séquençage d'ARN de cellules uniques dans un espace sémantique de faible dimension, générant des variables sémantiques (Z_sem) contenant des informations sur le type cellulaire et les perturbations. Dans le cadre de la recherche pharmaceutique, cet encodeur intègre des empreintes digitales de classe fonctionnelle recalibrées (r_FCFP), codant la structure moléculaire du médicament sous la forme d'un vecteur de dimension 2 048 intégré à l'espace sémantique. Afin de prédire les perturbations médicamenteuses inconnues, le modèle inclut également un module d'adaptation permettant la saisie de chaînes SMILES et d'informations posologiques, assurant ainsi une fusion poussée des informations biologiques et chimiques.
Le module DDIM conditionnel suit une conception à double processus de diffusion directe (diffusion dans l'espace génétique) et de diffusion inverse (diffusion inverse dans l'espace génétique).Au cours du processus de diffusion directe, les données d'expression génétique originales (x₀) sont progressivement transformées en un bruit approximativement pur (x₀) à travers 1 000 itérations.Dans ce processus, les trois types cellulaires typiques convergent progressivement vers une distribution gaussienne, tandis que Z_sem capture efficacement les variations biologiques de l'expression génique, séparant clairement les différentes conditions expérimentales dans l'espace latent. Lors de la rétrodiffusion, un réseau de prédiction du bruit doté d'un plongement de position sinusoïdal (ε) est utilisé.En utilisant l'étape de temps (t) et Z_sem comme conditions duales, le transcriptome biologiquement significatif a été reconstruit à partir de x_T par débruitage itératif, restaurant avec succès le profil de transcriptome original.
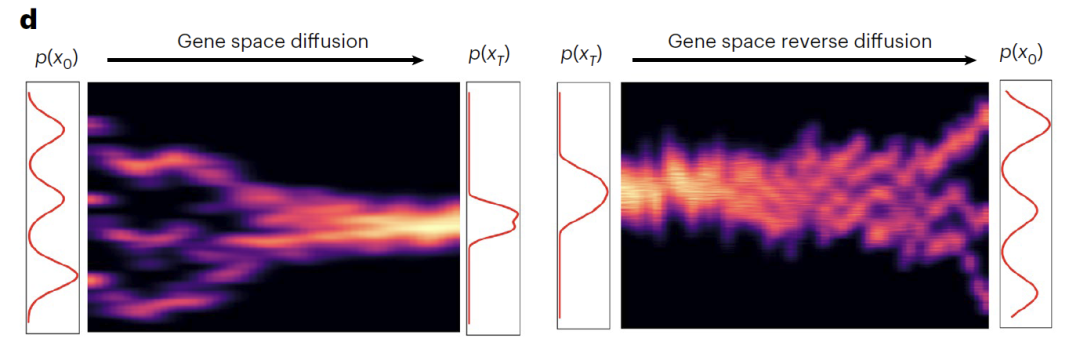
L'entraînement du modèle se concentre sur la perte de prédiction du bruit comme objectif d'optimisation principal, utilise l'optimiseur Adam (taux d'apprentissage 1×10⁻⁴) et s'appuie sur l'accélération GPU.En coordonnant la régulation des intervalles de temps et des variables sémantiques, le modèle peut simuler l'évolution continue des états cellulaires, fournissant ainsi un support pour la prédiction dynamique des trajectoires.
Comparé aux auto-encodeurs variationnels traditionnels, Squidiff présente des avantages significatifs :Sans nécessiter l'hypothèse d'une distribution gaussienne, ce modèle capture des profils d'expression génique complexes grâce à une réduction fine du bruit, améliorant le score F1 de 27% dans la prédiction de types cellulaires rares (<5%). Il introduit de manière novatrice la stratégie d'« interpolation de gradient », qui génère des trajectoires de différenciation continues en combinant linéairement des variables sémantiques dans l'espace latent, et identifie avec succès des états cellulaires transitoires souvent négligés par les modèles traditionnels (tels que les précurseurs mésodermiques lors de la différenciation des cellules iPS).
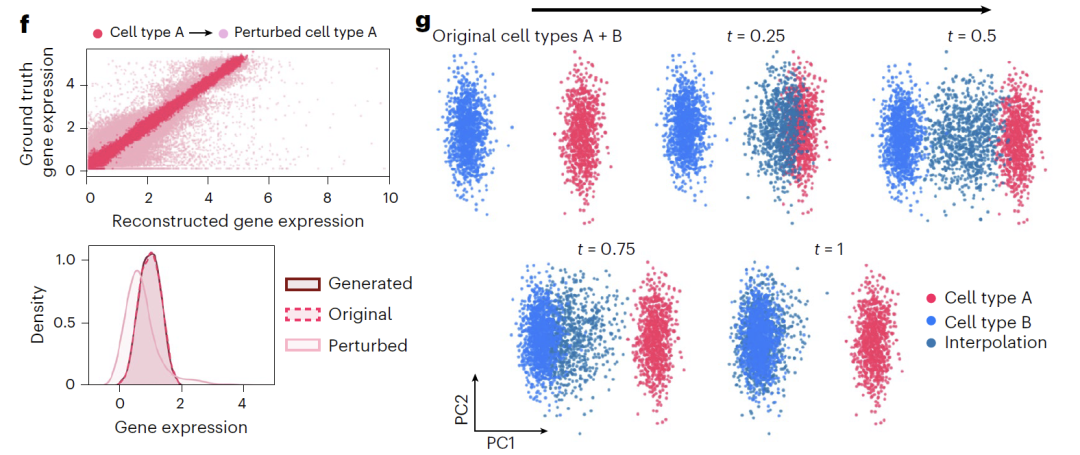
En outre, le modèle propose deux méthodes de manipulation des variables latentes : « addition » combine la représentation originale avec la direction de perturbation (Δz_sem), comme illustré dans la figure f ci-dessous, pour décaler la distribution de l’expression génique et refléter l’effet de la perturbation ; « interpolation » utilise l’interpolation linéaire, comme illustré dans la figure g ci-dessous, pour générer des états continus en obtenant des points intermédiaires sur la ligne de connexion vectorielle, réalisant ainsi une transition en douceur des types cellulaires.
Démonstration multi-scénarios de Squidiff : Capture précise des changements transcriptomiques lors de la différenciation cellulaire, des perturbations et de la réponse aux radiations
Afin de vérifier systématiquement les capacités de prédiction du transcriptome de Squidiff, l'équipe de recherche a mené une vérification expérimentale dans quatre domaines clés : la différenciation cellulaire, la perturbation des gènes et des médicaments, le développement des organoïdes vasculaires et les dommages causés par les radiations.
Pour la prédiction de la différenciation cellulaire, comme illustré ci-dessous, l'équipe a entraîné le modèle en utilisant uniquement les données des jours 0 et 3 de l'ensemble de données de différenciation des cellules iPSC en endoderme. La direction de la différenciation a été obtenue par le calcul des différences entre variables sémantiques, et Squidiff a prédit avec succès l'état intermédiaire entre les jours 1 et 2. Le modèle a reproduit fidèlement la diminution de l'expression du marqueur de pluripotence MMOG, la surexpression du facteur endodermique GATA6 et a identifié l'expression transitoire du marqueur mésodermique DBX1. Comparées aux méthodes traditionnelles, les données transcriptomiques générées par Squidiff permettent de reconstituer une trajectoire continue très cohérente avec la trajectoire de développement réelle.
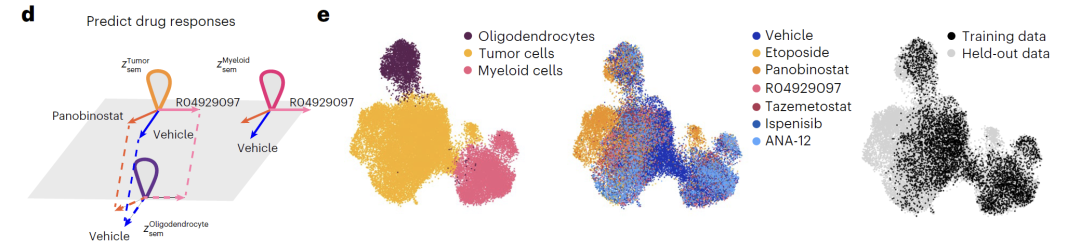
Le modèle démontre des performances exceptionnelles dans la prédiction des perturbations génétiques et médicamenteuses.Pour les expériences de double invalidation de gènes dans les cellules K562, Squidiff peut prédire avec précision les effets non additifs sans connaissance préalable, et sa robustesse surpasse les méthodes existantes.Lors d'essais cliniques, le modèle a permis de prédire les effets synergiques de combinaisons médicamenteuses à partir de données obtenues avec un seul médicament et d'identifier avec précision les effets spécifiques du pabicept sur les cellules tumorales. De plus, grâce à l'intégration d'un adaptateur de composés médicamenteux, ses performances prédictives pour le médicament inconnu SGLT1 étaient comparables à celles de modèles spécialisés, démontrant ainsi une excellente capacité de généralisation.
Dans leurs recherches sur les organoïdes vasculaires (BVO), l'équipe a réussi à prédire les états cellulaires à plusieurs points temporels intermédiaires en utilisant un modèle BVO induit par iPSC.Le modèle a non seulement reproduit les trajectoires de différenciation des trois principaux types cellulaires (cellules endothéliales, fibroblastes et cellules murales), mais a également identifié l'état intermédiaire de la différenciation des cellules murales en cellules endothéliales, difficile à observer par les méthodes traditionnelles. L'analyse de l'expression génique a montré que les modifications génétiques caractéristiques des données prédites étaient très cohérentes avec les profils de développement connus.
Dans les études sur les dommages radio-induits, le modèle a prédit avec précision les effets des radiations sur différents types cellulaires à partir de données d'apprentissage issues uniquement de cellules endothéliales. L'analyse a montré que les cellules à développement précoce étaient plus sensibles aux radiations, et les gènes différentiellement exprimés ainsi que les voies de signalisation associées prédites par le modèle ont été confirmés expérimentalement. Concernant la prédiction des effets protecteurs du G-CSF, le modèle a révélé les mécanismes protecteurs de ce médicament contre différents types cellulaires : activation des voies d'angiogenèse dans les fibroblastes, inhibition des voies d'apoptose dans les cellules endothéliales et amélioration de la stabilité génomique dans les cellules de la paroi. La validation expérimentale a montré une réduction significative de la mort cellulaire après traitement par G-CSF, démontrant ainsi la fiabilité des prédictions du modèle.
Ces expériences systémiques démontrent que Squidiff peut non seulement prédire avec précision les changements d'état cellulaire dans divers scénarios biologiques, mais aussi capturer les états transitoires et déduire les perturbations inconnues, fournissant ainsi un outil informatique puissant et fiable pour prédire les réponses cellulaires.
Un nouveau paradigme piloté par l'IA pour la recherche unicellulaire
Dans le domaine interdisciplinaire de la biologie unicellulaire et de l'intelligence artificielle, la percée technologique en matière de modélisation de la diffusion que représente Squidiff stimule l'innovation collaborative entre le monde universitaire et l'industrie.
Au niveau de la recherche universitaire, les meilleures équipes universitaires du monde entier continuent de réaliser des percées dans la profondeur et l'étendue de la modélisation unicellulaire.Une équipe de recherche de l'Université de Toronto, au Canada, a développé et publié scGPT, le premier modèle de langage fondamental à grande échelle pour la biologie unicellulaire.Ce modèle repose sur une architecture Transformer générative pré-entraînée et a été entraîné sur plus de 33 millions de points de données cellulaires couvrant 51 organes et tissus humains et 441 études indépendantes. Il couvre de manière exhaustive de multiples types cellulaires ainsi que des états physiologiques et pathologiques, et présente un atlas complet de l'hétérogénéité cellulaire humaine.
Titre de l'article :scGPT : Vers la construction d’un modèle de base pour l’analyse multi-omique unicellulaire à l’aide de l’IA générative
Adresse du document :
https://biorxiv.org/content/10.1101/2023.04.30.538439
en même temps,L'équipe de l'université de Stanford s'est concentrée sur l'innovation dans la dimension spatiale, en développant le cadre de modélisation spatio-temporelle tridimensionnel Spateo.S’appuyant sur des algorithmes évolutifs et précis, ce cadre peut reconstruire des modèles complets d’embryons et d’organes en trois dimensions à partir de données de coupes de tissus bidimensionnelles continues, et construire un système numérique spatial à plusieurs niveaux, des caractéristiques moléculaires unicellulaires à la morphologie macroscopique de l’embryon.
Titre de l'article :Modélisation spatio-temporelle des hologrammes moléculaires
Adresse du document :
https://www.cell.com/cell/fulltext/S0092-8674(24)01159-0
Le monde des affaires transforme ces découvertes universitaires en outils pratiques, démontrant ainsi leur valeur significative dans le développement de médicaments, le traitement des maladies et d'autres domaines.Cell2Sentence-Scale 27B (C2S-Scale 27B), développé par Google en collaboration avec l'université de Yale et d'autres institutions, est l'un des plus grands modèles de base au monde pour l'analyse unicellulaire.Ce modèle, basé sur la famille de modèles open source Gemma, possède 27 milliards de paramètres et permet une analyse approfondie des profils d'expression génique dans les cellules individuelles, ainsi que la prédiction précise des réponses cellulaires aux traitements médicamenteux. Actuellement intégré à la plateforme de criblage de médicaments de Google Health, il contribue à la conception de thérapies combinatoires personnalisées pour les tumeurs peu sensibles et accélère le développement de protocoles d'immunothérapie.Une autre pratique importante découle de la collaboration entre l'institut à but non lucratif Arc Institute et des entreprises telles que 10x Genomics, dont le modèle STATE se concentre sur la simulation des réponses cellulaires dynamiques.Il intègre des données d'observation provenant de 170 millions de cellules et des données d'intervention provenant de 100 millions de cellules, permettant une simulation précise des changements transcriptomiques dans les cellules soumises à un traitement médicamenteux, à une édition génique ou à une exposition aux radiations.
Il n’est pas difficile de constater que, depuis l’exploration approfondie des modèles unicellulaires de base par la communauté universitaire jusqu’à la mise en œuvre à grande échelle de cette technologie par l’industrie,La technologie de modélisation de la diffusion de Squidiff fait passer la recherche unicellulaire de « l'analyse de l'état cellulaire » à la « prédiction du devenir cellulaire ».Ce bond en avant accélère non seulement les progrès dans des domaines tels que le développement de médicaments et le traitement du cancer, mais fournira également un soutien technologique essentiel aux futures orientations médicales telles que la médecine de précision et la médecine régénérative, libérant ainsi en permanence l'énorme potentiel de l'innovation pilotée par l'IA dans les sciences de la vie.
Articles de référence :
1.https://mp.weixin.qq.com/s/yCR_GC0Ln80st2tHcv08-Q
2.https://mp.weixin.qq.com/s/GegQB65w4nZG6ZXvnyU9dw








