Command Palette
Search for a command to run...
Sélectionné Pour l'AAAI 2025 ! L'Université Tsinghua/UCL a Été La Pionnière d'une Solution De Fusion De Modèles De Langage Protéine-arn, Combinant La Prédiction d'affinité Pour Rafraîchir SOTA
Maladie d'Alzheimer, maladie de Parkinson, épilepsie... Ces maladies neurodégénératives « notoirement terrifiantes » sont des tueurs invisibles de la santé des personnes âgées, et l'apparition de ces maladies est souvent liée à la liaison anormale entre protéines et ARN.
Dans le domaine biomédical, l’étude de la liaison protéine-ARN est d’une importance vitale car elle joue un rôle central dans de nombreux processus biologiques tels que la régulation de l’expression des gènes, le traitement et l’épissage de l’ARN, la régulation de la traduction et la réponse au stress cellulaire.Comprendre le mécanisme de liaison protéine-ARN est la clé pour révéler des processus complexes de régulation des gènes et analyser la base génétique des maladies. Dans le même temps, les interactions protéine-ARN ont également des applications importantes dans la thérapie ciblée par l’ARN, ouvrant de nouvelles perspectives pour le traitement du cancer, des maladies génétiques et des maladies virales.
Récemment, parmi les réalisations sélectionnées annoncées lors de la 39e conférence annuelle de l'AAAI sur l'intelligence artificielle (AAAI 2025), la plus importante conférence internationale sur l'intelligence artificielle,Une équipe conjointe de l'Université Tsinghua, de l'University College London, de l'Université Monash et de l'Université des postes et télécommunications de PékinproposerLe modèle CoPRA a attiré une large attention dans l’industrie et a été sélectionné pour l’étape orale.
Il s'agit de la première tentative de combiner le modèle de langage protéique (PLM) et le modèle de langage ARN (RLM) via une architecture structurelle complexe pour la prédiction de l'affinité de liaison protéine-ARN.Pour tester les performances de CoPRA, les chercheurs ont compilé le plus grand ensemble de données d'affinité de liaison protéine-ARN à partir de plusieurs sources de données et ont évalué les performances du modèle sur 3 ensembles de données. Les résultats ont montré que CoPRA a atteint des performances de pointe sur plusieurs ensembles de données.
Les résultats associés sont intitulés « CoPRA : Bridging Cross-domain Pretrained Sequence Models with Complex Structures for Protein-RNA Binding Affinity Prediction » et ont été publiés sous forme de pré-impression sur arXiv.

Adresse du document :
https://arxiv.org/abs/2409.03773
Adresse de l'entrepôt CoPRA :
https://github.com/hanrthu/CoPRA
Le projet open source « awesome-ai4s » rassemble plus de 200 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
La recherche biomédicale continue de faire progresser les interactions protéines-ARN
Au cours des dernières années, les chercheurs du domaine biomédical n’ont cessé d’étudier les interactions protéines-ARN et ont réalisé des progrès considérables.
La technologie expérimentale CLIP est l’une des technologies les plus importantes dans la recherche sur l’ARN. Il peut analyser la carte de liaison de la protéine de liaison à l'ARN (RBP) sur l'ensemble du transcriptome et constitue la base de la compréhension systématique de la fonction d'une RBP et de son mécanisme de régulation. Cependant, les expériences CLIP prennent du temps et demandent beaucoup de travail, et ne peuvent fournir le site de liaison de l'ARN d'un certain RBP que dans un environnement cellulaire spécifique à la fois, et ont des exigences élevées en matière de matériel expérimental. Cependant, la liaison des protéines et de l’ARN peut changer considérablement avec les changements de l’environnement cellulaire, mais l’étude de la régulation des protéines sur l’ARN nécessite des informations de liaison dans le même environnement cellulaire.
Afin de résoudre le problème des changements dynamiques dans la liaison RBP dans différents environnements cellulaires,En février 2021, le groupe de recherche de Zhang Qiangfeng au Centre d'innovation avancée en biologie structurelle de l'Université Tsinghua a publié un résultat de recherche intitulé « Prédire les interactions dynamiques protéines-ARN cellulaires par apprentissage profond à l'aide de structures d'ARN in vivo » dans la revue Cell Research. Ce travail a utilisé l'expérience icSHAPE pour analyser les cartes de structure secondaire de l'ARN de 7 types de cellules courantes, et a développé un algorithme d'intelligence artificielle pour intégrer la structure de l'ARN intracellulaire obtenue à partir de l'expérience et les informations de liaison RBP de l'environnement cellulaire correspondant, et a établi une nouvelle méthode PrismNet pour prédire la liaison dynamique du RBP intracellulaire sur la base des informations sur la structure de l'ARN intracellulaire.
Afin de prédire l’affinité de liaison protéine-ARN, plusieurs méthodes de calcul ont été proposées dans l’industrie.Comprend des méthodes basées sur la séquence et sur la structure. Les méthodes basées sur les séquences traitent les séquences de protéines et d'ARN séparément à l'aide de différents encodeurs de séquence et modélisent ensuite les interactions entre elles. Cependant, les performances de ces approches sont souvent limitées car l’affinité de liaison est principalement déterminée par la structure de l’interface de liaison. D’autres méthodes récemment proposées se concentrent sur l’extraction des caractéristiques structurelles de l’interface de liaison, telles que l’énergie et la distance de contact. Sur la base de ces caractéristiques extraites, les chercheurs ont développé une approche d’apprentissage automatique basée sur la structure pour la prédiction d’affinité. Cependant, en raison de la limitation de la taille de l’ensemble de données, ces méthodes ont une capacité de généralisation limitée sur de nouveaux échantillons et dépendent fortement de l’ingénierie des caractéristiques.
Avec l’essor de la technologie de l’intelligence artificielle, de nombreux modèles de langage protéique (PLM) et modèles de langage ARN (RLM) ont été développés, qui ont démontré d’excellentes performances et capacités de généralisation dans diverses tâches en aval.Dans le même temps, la structure tridimensionnelle des protéines/ARN étant essentielle à la compréhension de leurs fonctions, l’intégration d’informations structurelles dans les modèles linguistiques est également devenue une nouvelle tendance.
Par exemple, une équipe de l’Université du Missouri, de l’Université du Kentucky et de l’Université de l’Alabama a utilisé une technologie d’apprentissage comparatif multi-perspectives pour intégrer des informations clés sur la structure des protéines dans le modèle de langage des protéines. Sur la base de ce concept, l’équipe a développé S-PLM : un modèle de langage protéique capable de percevoir les informations structurelles 3D des protéines. S-PLM démontre d'excellentes performances dans de multiples tâches de prédiction de protéines. Après une formation à l'aide d'un outil de réglage léger, les performances de S-PLM atteignent ou dépassent les méthodes de pointe actuelles dans des tâches telles que la prédiction de la fonction des protéines, la prédiction de la classe de réaction enzymatique et la prédiction de la structure secondaire. La recherche connexe a été publiée sur bioRxiv sous le titre « S-PLM : Structure-aware Protein Language Model via Contrastive Learning between Sequence and Structure ».
Cependant, bien que les recherches industrielles actuelles aient démontré le grand potentiel des modèles de langage biologique pilotés par des informations structurelles dans les tâches interactives, les travaux combinant des modèles pré-entraînés issus de différents domaines biologiques sont encore rares.Dans CoPRA, proposé conjointement par l'Université Tsinghua, l'University College London, l'Université Monash et l'Université des Postes et Télécommunications de Pékin, une tentative a été faite pour la première fois pour combiner des modèles de langage de protéines et d'ARN avec des informations structurelles complexes pour la prédiction de l'affinité de liaison protéine-ARN.
Conception d'un modèle Co-Former léger pour construire CoPRA
Dans l’ensemble, le processus de construction du modèle CoPRA est illustré dans la figure suivante :
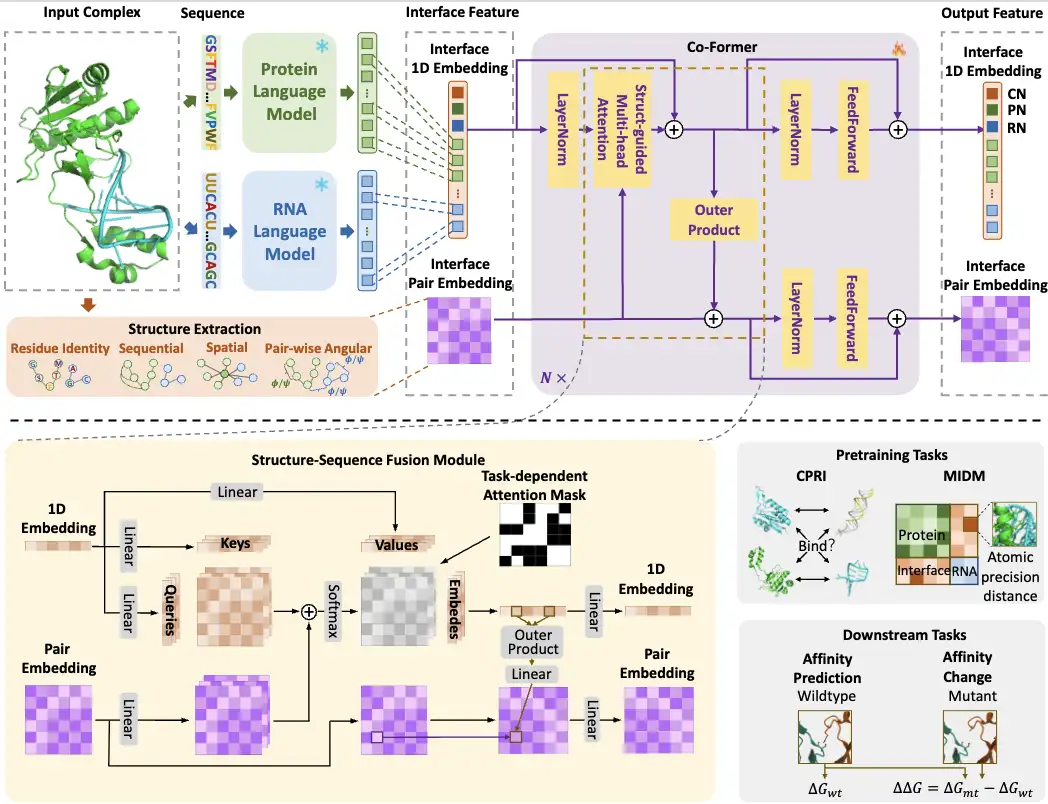
d'abord,Les chercheurs ont introduit des séquences de protéines et d'ARN dans PLM et RLM respectivement, puis ont sélectionné les intégrations à l'interface d'interaction à partir des sorties des deux modèles de langage comme intégrations de séquences pour l'apprentissage intermodal ultérieur. Dans le même temps, il extrait également des informations structurelles (caractéristiques d'interface) de l'interface d'interaction sous forme d'intégration appariée.
Alors,Les chercheurs ont conçu un modèle Co-Former léger qui combine des intégrations de séquences d'interface provenant de deux modèles de langage avec des informations structurelles complexes pour former un module de fusion structure-séquence. Plus précisément, Co-Former fusionne les intégrations 1D et par paires via des modules d'auto-attention multi-têtes guidés par la structure et des modules de produits externes, et applique des masques d'attention dépendants de la tâche. Les nœuds spéciaux de sortie et les plongements appariés de Co-Former sont utilisés selon différentes tâches, dont deux tâches de pré-formation et deux tâches d'affinité en aval.
Les chercheurs ont également proposé une stratégie de pré-entraînement à double portée pour Co-Former.Pour modéliser la classification des interactions contrastives à gros grains (CPRI) et la prédiction de la distance d'interface à grains fins (MIDM), apprises avec une précision au niveau atomique.
Pour évaluer les performances de CoPRA et d'autres modèles,Les chercheurs doivent remédier au manque d’ensembles de données standard d’annotation unifiés. Ils ont donc collecté des échantillons à partir de trois ensembles de données publics : PDBbind, PRBABv2 et ProNAB, compilé le plus grand ensemble de données d'affinité de liaison protéine-ARN PRA310 et évalué la capacité de leur modèle à prédire l'affinité de liaison protéine-ARN sur les ensembles de données PRA310 et PRA201.
*Ensemble de données PRA201 : un sous-ensemble de PRA310, chaque complexe contient une seule chaîne protéique et une seule chaîne d'ARN, et a des contraintes de longueur plus strictes
CoPRA est le meilleur outil pour prédire l'affinité de liaison protéine-ARN
Comme le montre le tableau ci-dessous, la version formée de novo de CoPRA obtient les meilleures performances sur l'ensemble de données PRA310. De plus, la plupart des méthodes utilisant des intégrations LM comme entrée surpassent les autres méthodes, indiquant le grand potentiel de la combinaison de LM unimodaux pré-entraînés pour la prédiction d'affinité.
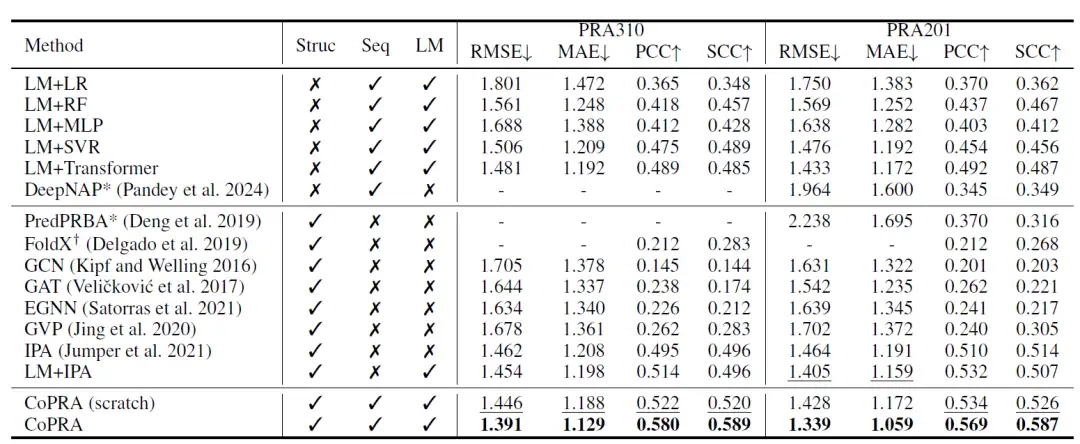
Les chercheurs ont ensuite pré-entraîné le modèle à l’aide de leur ensemble de données compilé non supervisé PRI30k, améliorant considérablement ses performances globales sur les deux ensembles de données. Sur l'ensemble de données PRA310, CoPRA atteint un RMSE de 1,391, un MAE de 1,129, un PCC de 0,580 et un SCC de 0,589, ce qui est bien meilleur que le deuxième meilleur modèle CoPRA (entraîné à partir de zéro). PredPRBA et DeepNAP prennent en charge la prédiction d'affinité des paires protéine-ARN. Les chercheurs ont comparé les performances de ces méthodes sur l'ensemble de données PRA201 et ont montré que leurs performances sur PRA201 étaient significativement inférieures à leurs résultats rapportés, même si au moins 100 échantillons dans PRA201 apparaissaient dans leurs ensembles d'entraînement, indiquant que ces méthodes ont de faibles capacités de généralisation.
CoPRA est plus efficace pour prédire l'effet des mutations sur l'affinité de liaison et possède une excellente capacité de généralisation
Pour évaluer davantage la compréhension fine de l’affinité du modèle, les chercheurs ont redirigé le modèle pour prédire les effets des mutations ponctuelles de la protéine sur le complexe protéine-ARN. En se référant à des études connexes sur la prédiction de l'effet de la mutation des protéines, les chercheurs ont fait la moyenne des mesures à chaque niveau complexe et ont évalué les performances de tir zéro et de réglage fin de CoPRA après pré-formation sur PRI30k et réglage sur PRA310.
Comme le montre le tableau ci-dessous, après un réglage précis à l'aide de l'ensemble de validation croisée du mCSM, le modèle proposé dans cette étude a surpassé les autres modèles dans les quatre indicateurs, avec un RMSE de 0,957, un MAE de 0,833, un PCC de 0,550 et un SCC de 0,570.
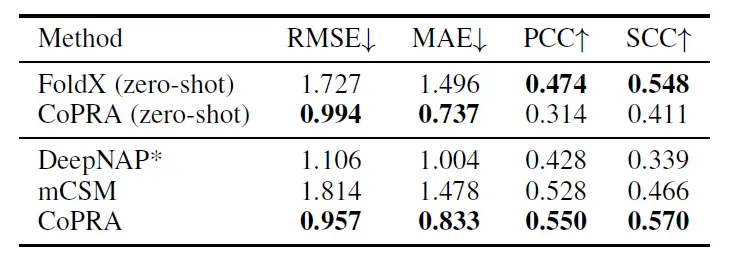
Cette performance supérieure découle du double objectif de pré-formation malgré l'absence de structures complexes mutantes, démontrant la capacité de généralisation de CoPRA sur différentes tâches liées à l'affinité.
Progrès révolutionnaires dans les modèles de langage protéique multimodal
L’essence de l’idée de recherche introduite ci-dessus est de combiner plusieurs modalités biologiques telles que les protéines et l’ARN avec des informations structurelles complexes, ce qu’on appelle l’apprentissage multimodal. En termes simples, l’apprentissage multimodal est le processus d’intégration de différents types de données dans un seul modèle dans le cadre de l’apprentissage profond.
Au cours des dernières années, avec le développement rapide de grands modèles linguistiques, les chercheurs ont commencé à essayer de les appliquer au domaine de la science des protéines pour comprendre et prédire avec précision la fonction, la structure et les propriétés des protéines. Cependant, les modèles de langage à grande échelle orientés vers les protéines traitent principalement les séquences d’acides aminés comme du texte, sans parvenir à exploiter pleinement les riches informations structurelles des protéines.Aujourd’hui, les progrès dans l’apprentissage multimodal ont fourni de nouvelles idées pour de plus en plus de recherches connexes.
Par exemple, dans le domaine de la recherche et du développement de médicaments, une prédiction précise et efficace de l’affinité de liaison entre les protéines et les ligands est cruciale pour le criblage et l’optimisation des médicaments. Cependant, les études précédentes n’ont pas pris en compte le rôle important des informations de surface moléculaire dans les interactions protéine-ligand. Sur cette base,Des chercheurs de l'Université de Xiamen ont proposé un nouveau cadre d'extraction de caractéristiques multimodales (MFE).Ce cadre combine pour la première fois les informations sur la surface des protéines, la structure 3D et la séquence, et utilise un mécanisme d'attention croisée pour aligner les caractéristiques entre différentes modalités. Les résultats expérimentaux montrent que cette méthode a atteint des performances de pointe dans la prédiction de l’affinité de liaison protéine-ligand. La recherche connexe a été publiée dans Bioinformatics en juin 2024 sous le titre « Prédiction de l’affinité de liaison protéine-ligand multimodale basée sur la surface ».
En décembre 2024, une équipe de recherche de l'Université normale de Chine de l'Est et d'autres institutions a proposé une solution innovante, EvoLLama.Il s'agit d'un cadre qui intègre un encodeur de structure protéique, un encodeur de séquence et un grand modèle de langage pour la fusion multimodale. Dans le cadre du zero-shot, EvoLLama démontre de fortes capacités de généralisation, améliorant les performances d'autres modèles de base affinés de 1% à 8%, et dépassant les performances moyennes des modèles de réglage fin supervisés de pointe actuels de 6%. Les résultats de recherche pertinents ont été publiés sous forme de pré-impression sur arXiv sous le titre « EvoLlama : Améliorer la compréhension des protéines par les LLM via des représentations multimodales de structures et de séquences ».
Bien entendu, l’apprentissage multimodal n’est qu’une des options de recherche disponibles. À l’avenir, en utilisant davantage de méthodes d’apprentissage automatique pour étudier la surface des protéines, les biologistes pourront mieux comprendre comment elles interagissent avec d’autres molécules biologiques, contribuant ainsi au développement de nouveaux médicaments.
Références :
1.https://arxiv.org/abs/2409.03773
2.https://www.frcbs.tsinghua.edu.cn/index.php?c=show&id=873
3.https://www.sohu.com/a/846589543_121124715








