Command Palette
Search for a command to run...
Inspired by DeepSeek Engram, the "external Brain" of the Genome Basic Model, Gengram, Achieved a Performance Improvement of up to 22.61 TP3T.

Basic genome models (GFMs) are core tools for decoding the code of life, unlocking key biological information such as cell function and organism development by analyzing DNA sequences. However, existing Transformer-based GFMs have a fatal flaw: they rely on large-scale pre-training and intensive computation to indirectly infer polynucleotide motifs, which is not only inefficient but also limited in motif-driven functional element detection tasks.
recently,The Gengram (Genomic Engram) model proposed by the Genos team, comprised of members from BGI Life Sciences Research Institute and Zhejiang Zhijiang Laboratory,This provides a revolutionary solution to this problem. This design avoids hard-coding biological rules while giving the model an explicit understanding of the genomic "grammar".
As a lightweight conditional memory module designed specifically for genome motif modeling, Gengram's core innovation lies in its k-mer hash memory mechanism, which constructs a highly efficient multi-base motif memory repository. Unlike traditional models that indirectly infer motifs,It directly stores k-mers of 1-6 base lengths and their embedding vectors, and captures the local contextual dependencies of functional motifs through a local window aggregation mechanism.The motif information is then fused with the backbone network via a gate-controlled module. The research team stated that when integrated into the current state-of-the-art (SOTA) genome model Genos, under the same training conditions, Gengram achieves significant performance improvements across multiple functional genomics tasks, with a maximum improvement of 22.61 TP3T.

Paper address:https://arxiv.org/abs/2601.22203
Code address:https://github.com/BGI-HangzhouAI/Gengram
Model weights:https://huggingface.co/BGI-HangzhouAI/Gengram
Training data covers human and non-human primate genomes.
The training dataset contains 145 high-quality haplotype-parsed and assembled sequences, covering human and non-human primate genomes.Human sequences were primarily derived from the Human Pangenome Reference Consortium (HPRC, 2nd edition), supplemented by the GRCh38 and CHM13 reference genomes. Non-human primate sequences were integrated from the NCBI RefSeq database to incorporate evolutionary diversity. All sequences were processed using one-hot encoding. The vocabulary includes four standard bases (A, T, C, G), ambiguous nucleotides (N), and document end markers.
final,The system constructed three sets of data to support the ablation experiments and formal pre-training.
50B tokens @ 8,192 (ablation)
200B tokens @ 8k (10B formal pre-training)
100B tokens @ 32k (10B formal pre-training)
And maintain a data mixing ratio of human:non-human = 1:1.
Genome modeling is shifting from "attention derivation" to "memory enhancement".
Inspired by DeepSeek Engram's memory mechanism, the Genos team quickly developed and deployed Gengram.This module provides explicit motif storage and reuse capabilities for basic genome models, overcoming the limitations of mainstream GFMs, which lack structured motif memory and can only rely on expanding the "implicit memory" of training data. This drives genome modeling from "attention derivation" to "memory enhancement." The module architecture is shown in the figure below:
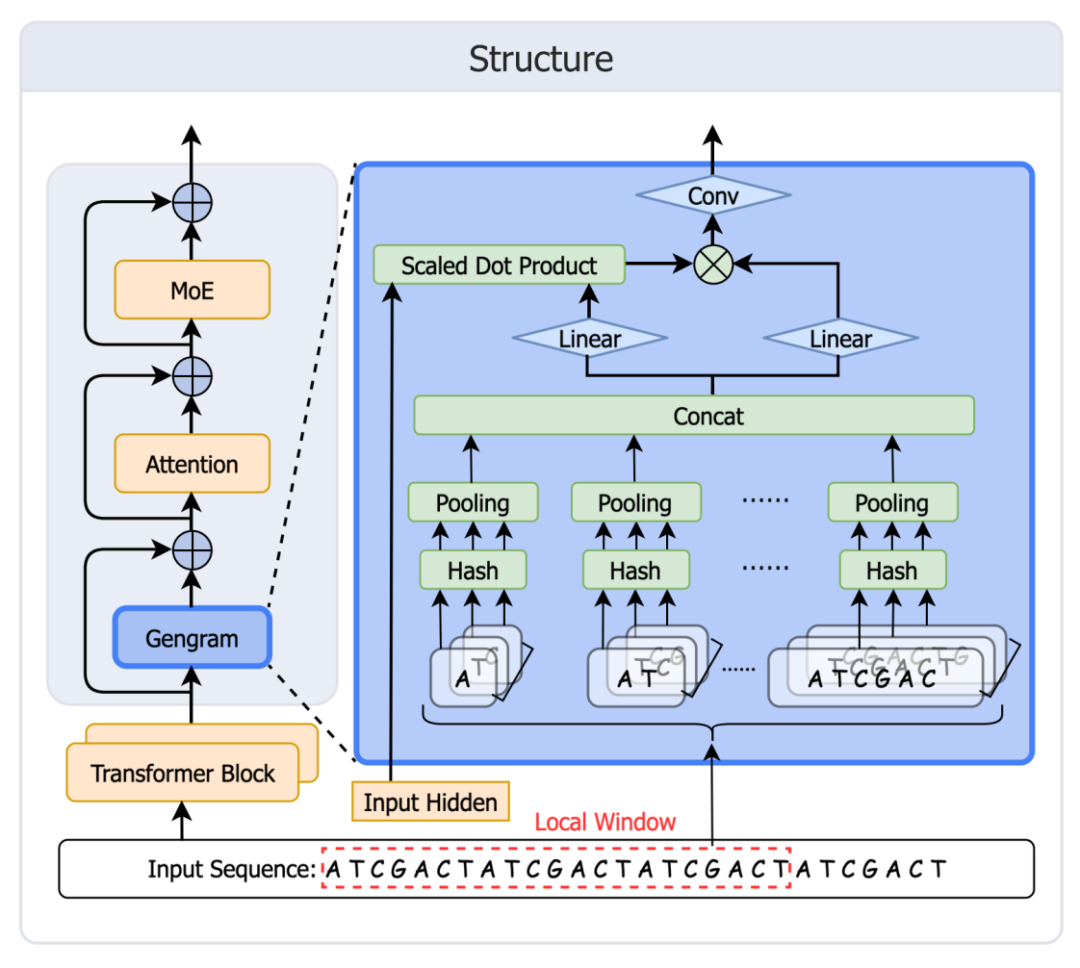
Table creation: Build a hash memory (static key + learnable embedding value) for all k-mer values from k=1 to 6.
Retrieval: Map all k-mer values appearing in the window to table entries.
Aggregation: First aggregate at each k, then concatenate across k.
Gating: The gate controls activation, writes motif evidence into the residual stream, and then enters attention.
A key design feature: Local Window Aggregation (W=21bp)
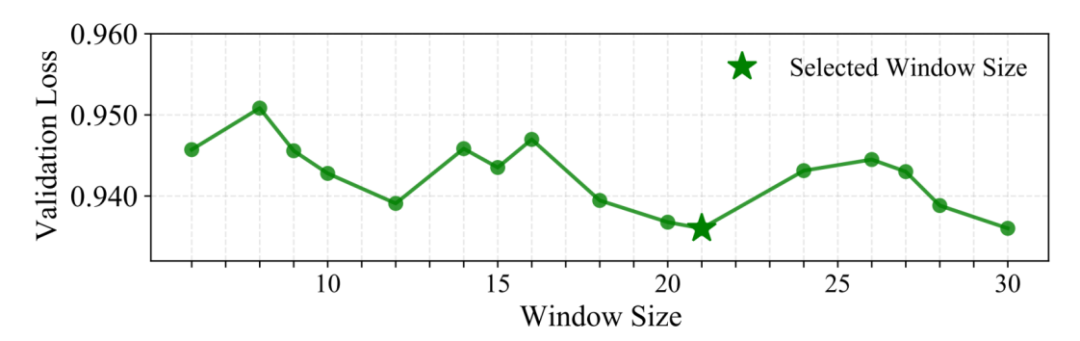
Instead of retrieving a single n-gram at each location, Gengram employs aggregation of multiple k-mer embeddings within a fixed window to more reliably inject "local, structurally consistent" motif evidence. Researchers validated this by searching using a window size strategy.We found that 21 bp achieves optimal performance on the validation set.One possible biological explanation is that the typical DNA double helix cycle is about 10.5 base pairs per rotation, so 21 base pairs rotate exactly twice. This means that two bases that are 21 bp apart are located on the same side of the helix in three-dimensional space, facing similar biochemical environments. Windowing at this scale may be more conducive to aligning the phase consistency of local sequence signals.
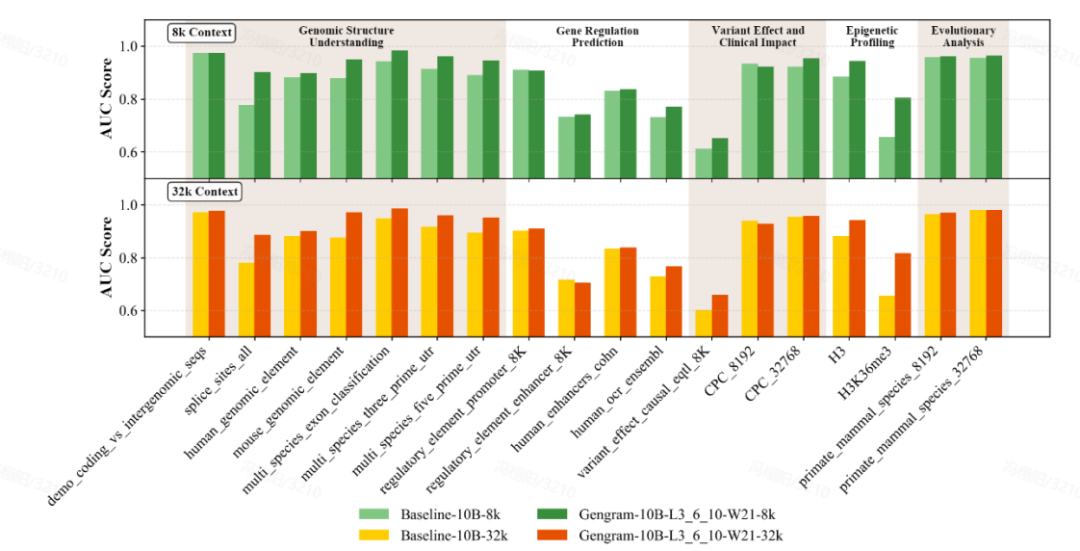
Significant Improvements in Evaluation: Small Parameters, Big Changes
The team conducted a comprehensive evaluation of the model using multi-standard benchmark datasets, covering Genomic Benchmarks (GB), Nucleotide Transformer Benchmarks (NTB), Long-Range Benchmarks (LRB), and Genos Benchmarks (GeB).Eighteen representative datasets were selected, covering five main task categories:Genomic Structure Understanding, Gene Regulation Prediction, Epigenetic Profiling, Variant Effect & Clinical Impact, and Evolutionary Analysis.
Gengram, a lightweight plugin with only about 20 million parameters, represents a tiny fraction of the parameters in a base model with hundreds of billions of parameters, yet it delivers significant performance improvements. Under the same training conditions, with context lengths of 8k and 32k...Models integrated with Gengram outperformed the unintegrated versions in the vast majority of tasks.In terms of specific manifestationsThe AUC score for the splice site prediction task improved from 0.776 to 0.901, an increase of 16.11 TP3T;The AUC score of the epigenetic prediction task (H3K36me3) improved from 0.656 to 0.804, an increase of 22.61 TP3T.
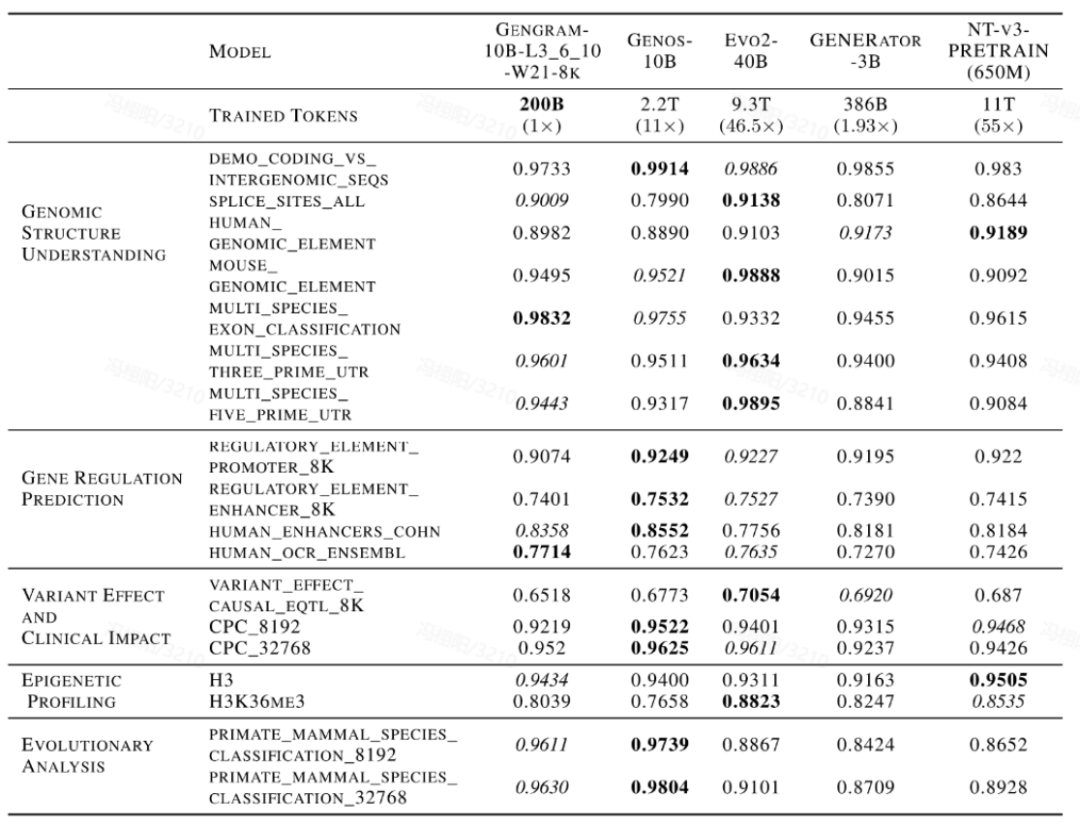
Furthermore, this performance improvement is accompanied by a significant "data leverage" effect. In a horizontal comparison with mainstream DNA-based models such as Evo2, NTv3, and GENERATOR-3B,Models integrating Gengrams require only a very small amount of training data and fewer activation parameters to rival publicly available models with training data that are several to tens of times larger on core tasks.It demonstrates high data training efficiency.
In-depth analysis of Gengram
Why can Gengram accelerate training?
The team introduced KL divergence as a representational diagnostic metric for the training process and used LogitLens-KL to quantify and track the "prediction-readiness" of different layers. The results showed that...By introducing Gengrams, the model can form a stable prediction distribution earlier in the shallow layers:Compared to the baseline model, its inter-layer KL values decrease faster and enter the low-value range earlier, indicating that effective supervision signals are organized into usable representations earlier, thus making gradient updates more direct and optimization paths smoother, ultimately resulting in faster convergence speed and higher training efficiency.
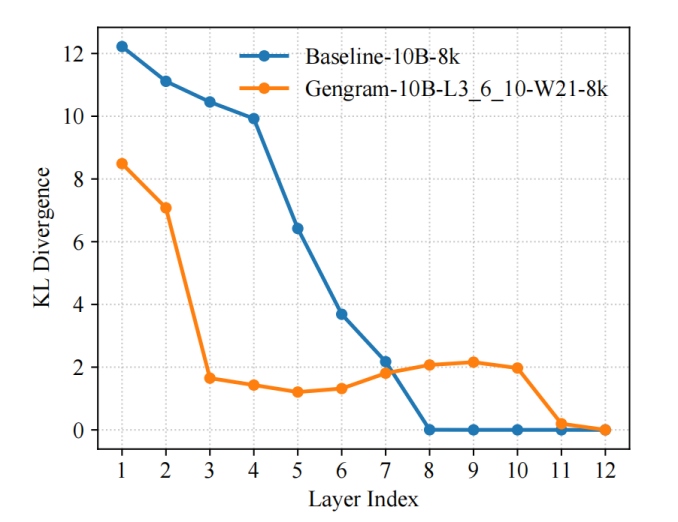
This phenomenon did not occur out of thin air, but was directly driven by Gengram's structural design:
Explicit motif memory retrieval shortens the path from evidence to representation. In genomic tasks, supervisory signals are often triggered by short and sparse motifs (such as spliced consensus sequences, promoter-related fragments, low-complexity tracts, etc.). Baseline Transformers need to gradually "derive and solidify" these local evidences through multiple layers of attention/MLP; while Gengrams, through explicit access to k-mers, directly provide these high-information-density local patterns to the network in the form of memory, so that the model does not have to wait for deep layers to gradually form motif detectors, and is closer to a predictable state from the beginning.
Window aggregation and dynamic gating make the injected evidence "stable and controllable". Gengrams do not perform position-by-position hard injection; instead, they aggregate multiple k-mer embeddings within a fixed window.Furthermore, gated selective writing to the residual stream is employed: retrieval is more likely to be activated in functional regions and suppressed in large background regions. This "sparse, aligned functional element" writing method reduces noise interference on the one hand, and allows the network to obtain high signal-to-noise ratio training signals earlier, thus reducing the optimization difficulty.
Where do Motif memories come from? A detailed explanation of Gengram's writing mechanism.
The research team first observed a clear and consistent phenomenon across tasks in downstream evaluations:Under the same training settings, the introduction of Gengrams significantly improved the model on typical motif-driven tasks, especially in scenarios relying on short program sequences, such as splice site identification and epigenetic histone modification site prediction. For example, on representative tasks, the AUC for splice site prediction increased from 0.776 to 0.901, and the AUC for H3K36me3 prediction increased from 0.656 to 0.804, showing stable and substantial gains.
To further answer the question "Where do these improvements come from?", the team did not stop at the metric level, but extracted the residual writes of the Gengram from the model's forward propagation and visualized their intensity distribution in the sequence dimension as a heatmap for analysis.The results show that the written signal exhibits a highly sparse and high-contrast structure: most locations are close to the baseline, and only a few locations form sharp peaks;More importantly, these peaks are not random, but are significantly enriched and aligned with functionally relevant regions and boundaries, including TATA-box fragments near promoters, low-complexity poly-T fragments, and key locations near the boundaries of functional regions such as genes/exons.This means that writing to a Gengram is more like "grasping local evidence of its decisive function" rather than indiscriminately injecting information across the entire sequence.
Based on the above phenomena and the chain of evidence,Researchers can summarize Gengram's motif memory mechanism as "on-demand retrieval—selective writing—structured alignment":The module controls the intensity of retrieval and writing by gating, more actively injecting reusable motif evidence in regions with higher functional information density, and suppressing writing in background regions to reduce noise interference. As a result, the model's mastery of motifs no longer mainly relies on the "implicit memory" brought by larger-scale data, but instead shifts to a structured ability to explicitly access and interpretably write representations.

Conclusion
In recent years, the field of genome modeling has been undergoing a key shift from "sequence statistical learning" to "structure-aware modeling".
Conditional motif memory mechanisms, exemplified by Gengram, reveal a technical path distinct from traditional intensive computing: by explicitly modeling multi-base functional motifs as retrievable structured memories, the model can achieve more efficient and stable utilization of functional information while maintaining general architectural compatibility.This approach has not only demonstrated significant performance advantages in multiple functional genomics tasks, but also provided a unified engineering solution for sparse computation, long sequence modeling, and model interpretability.
Furthermore, from an industry perspective, the "structured prior + modular enhancement" paradigm embodied by Gengram significantly reduces the marginal costs of large-scale genome models in terms of computing power, data, and training cycles, making its large-scale deployment in high-value scenarios such as drug development, variant screening, and gene regulation analysis practically feasible. Looking further ahead, these reusable and pluggable architectural components may become the standard configuration for next-generation genome foundation models, driving the industry from "larger models" to "smarter models" and accelerating the continuous transformation of academic research results into industrial platforms and clinical applications.








