Command Palette
Search for a command to run...
With an Accuracy of 971 TP3T! Princeton University and Others Proposed MOFSeq-LMM, Which Efficiently Predicts Whether MOFs Can Be synthesized.
Metal-organic frameworks (MOFs) have shown great potential in applications such as gas storage, separation, catalysis, and drug delivery due to their highly tunable pore structures and rich chemical functionalities. However,MOFs have a vast design space encompassing trillions of possible combinations of building blocks, making experimental exploration extremely inefficient.
To accelerate the discovery of MOFs, computational pipelines have emerged, aiming to generate novel MOFs, predict their properties, and ultimately achieve their synthesis. In this process,The main challenge lies in the low conversion rate from screening to synthesis.This largely stems from the uncertainty surrounding the feasibility of synthesizing computer-generated MOFs. For example, of the thousands of computational MOF screenings published to date, only about a dozen have been accompanied by MOF synthesis.
Free energy is an important indicator for evaluating the thermodynamic stability and synthesizability of MOFs, but traditional computational methods are costly on large-scale MOF datasets, making rapid screening difficult. To address this challenge, a joint research team from Princeton University and the Colorado School of Mines has proposed an efficient prediction method based on machine learning.By using large language models (LLM) to directly predict the free energy from the structural sequence of MOFs, computational costs can be significantly reduced, enabling high-throughput and scalable thermodynamic assessment of MOFs.The model exhibits high versatility without the need for retraining: its F1 score is as high as 97% when determining whether the free energy of MOFs is higher or lower than the empirically based synthetic feasibility threshold.
The related research findings, titled "Highly Accurate and Fast Prediction of MOF Free Energy via Machine Learning," have been published in ACS Publications.
Research highlights:
* Based on this model, researchers can predict free energy with high accuracy and simulate the results of complete molecular simulations without retraining, thereby determining the feasibility of synthesizing MOFs.
* Work that used to require a lot of time in the laboratory or through molecular simulations can now be done in a negligible amount of time.
This method provides a feasible approach for using machine learning free energy prediction as an early or late screening tool in performance-based computational MOF screening.

Paper address:
https://pubs.acs.org/doi/10.1021/jacs.5c13960
Follow our official WeChat account and reply "free energy prediction" in the background to get the full PDF.
More AI frontier papers:
MOFMinE: Covering 1 million MOF prototypes
To support model training,The research team constructed a massive MOFs dataset, MOFMinE, covering approximately one million MOF prototypes.It includes information on the entire process from component selection and topology template mapping to functional modifications, as shown in the following figure:
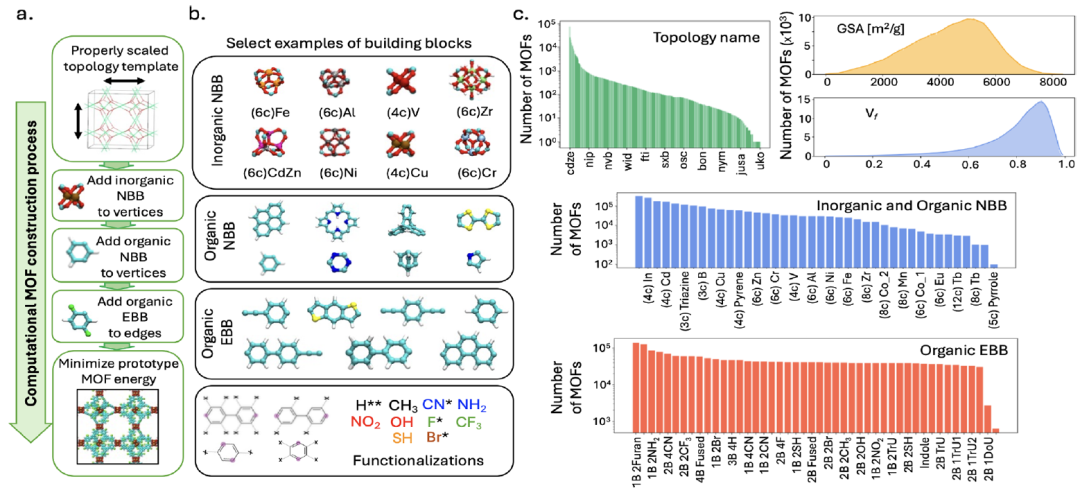
Construction method
The dataset is generated based on the ToBaCCo-3.0 platform. Each MOF is generated by mapping its constituent building units onto a topological template that has been appropriately scaled (to match the building unit size). This template guides the spatial arrangement and connectivity of the building units within the MOF unit cell. ToBaCCo building units are categorized into nodal building blocks (NBBs) or edge building blocks (EBBs) based on their mapping location: nodal building units are mapped to template vertices, and edge building units are mapped to template edges. NBBs can be inorganic or organic, with inorganic NBBs corresponding to the so-called MOF second-order building units (SBUs), and organic NBBs combining with EBBs to form MOF connectors.
Data scale and diversity
MOFMinE contains 1,393 topological templates, 27 inorganic NBBs, 14 organic NBBs, and 19 basic EBBs, and covers 13 functional modifications, ensuring diversity in chemical and topological structures.The database has a void fraction ranging from 0.01 to 0.99, a surface area (GSA) ranging from 26 to 8382 m²/g, and a maximum pore size (LPD) ranging from 2.6 to 127.7 Å, fully covering the structural space of MOFs.
Free energy subset
Of these 1 million MOF prototypes, a subset of 65,574 structures collected free energy data.This subset contains 379 topological templates, 6 inorganic NBBs, 11 organic NBBs, and 12 basic EBBs with 13 functionalization modifications. The porosity properties of the subset are: Vf ranging from 0.01 to 0.97, GSA ranging from 38 to 7304 m²/g, and LPD ranging from 2.6 to 87.8 Å. This dataset was used for free energy prediction fine-tuning and testing of LLMs.
MOFSeq-LMM model for efficient prediction of MOF free energy
With the support of the MOFMinE dataset,The research team constructed the MOFSeq-LMM model framework for efficiently predicting the free energy of MOFs and realizing data-driven design from structure to properties.The core idea of this framework is to transform the structural information of MOFs into a computer-understandable sequence representation (MOFSeq) and combine it with a large language model for learning and prediction, thereby significantly reducing computational costs while preserving physicochemical information.
MOFSeq characterization
To overcome the limitations of existing representation strategies and fully utilize large language models for extensive MOF property prediction,Researchers have developed MOFSeq. This novel string-based sequence representation method is both compact and highly informative, encoding the local and global structural features of MOFs in an optimized manner.This enables language models to be processed efficiently and scalably.
In MOFSeq, local information mainly includes the atomic composition of building blocks and their internal connectivity information; global information mainly includes high-level descriptions of MOF building blocks and the connection patterns between building blocks. Local information is obtained through the MOFid tool, while global information relies on ToBaCCo-3.0, as shown in the figure below:
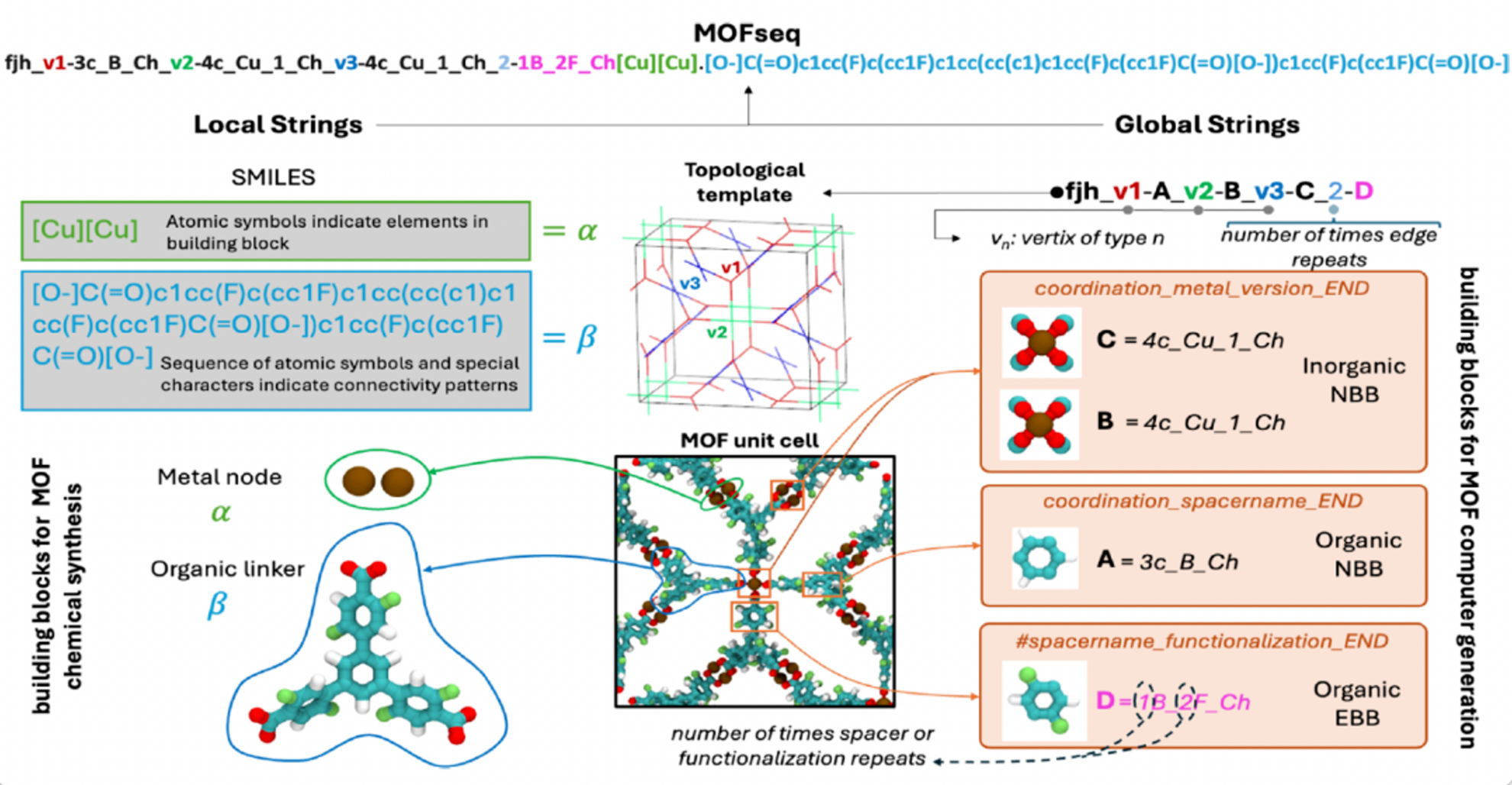
MOFs Database Construction and Data Processing
After constructing the MOFMinE dataset based on the method described above, all MOF prototypes generated by ToBaCCo were optimized using the UFF4MOF force field in LAMMPS (October 29, 2020 version) to obtain the final MOF structures.
The dataset generated using ToBaCCo-3.0 only contains the MOFname and its corresponding CIF file as a representation of each MOF. However, MOFSeq requires both the MOFname and MOFid.To obtain MOFid, the researchers used the MOFid generator developed by Bucior et al.This generator can generate MOFid and MOFkey simultaneously based on the CIF structure of MOF.
Ultimately, the 793,079 MOFSeq pre-training samples were divided into a training set of 634,463 samples, a validation set of 79,308 samples, and a test set of 79,308 samples. The 54,443 MOFSeq fine-tuning data points were divided into a training set of 43,554 samples, a validation set of 5,444 samples, and a test set of 5,445 samples.
LLM-Prop Model Design
Building upon MOFSeq characterization, the research team employed LLM-Prop, a large language model specifically designed for predicting material properties. The LLM-Prop model has a relatively moderate size, with approximately 35 million parameters, ensuring both learning ability and computational efficiency. The model input length is set to 2,000 tokens, which can accommodate the structural sequence information of most MOFs. Through an attention mechanism, the model can adaptively capture the influence of different components and topologies on the free energy in the sequence, forming an interactive representation of global and local features.
Pre-training and fine-tuning
* Pre-training phase:
Researchers trained LLM-Prop to predict the strain energies of MOFs using the MOFSeq representation. Strain energy was chosen because of its low computational cost and high correlation with free energy. Dropout rates of 0.2 and 0.5 were used during pre-training, with results showing that a dropout rate of 0.2 performed better in both pre-training and downstream tasks. The MOFSeq input length was set to 2000 tokens.
Fine-tuning phase:
The setup is the same as pre-training, but the model objective is changed to predicting free energy, and the number of training epochs is increased to 200. LLM-Prop is designed as a lightweight model, approximately 1/2000 the size of Llama 2, prioritizing computational efficiency. This design introduces a trade-off: compared to fine-tuning large LLMs (such as Llama 2 or GPT-2), LLM-Prop requires more training epochs to achieve high performance, but its small size makes training feasible and efficient.
The accuracy of predicting MOF synthesis reached 97%.
After training the MOFSeq-LMM model, the research team systematically evaluated its performance in free energy prediction, synthesis feasibility assessment, and polymorphic MOF screening. The experimental results not only validated the model's high accuracy but also highlighted its application potential in high-throughput MOF design and screening.
Free energy prediction performance
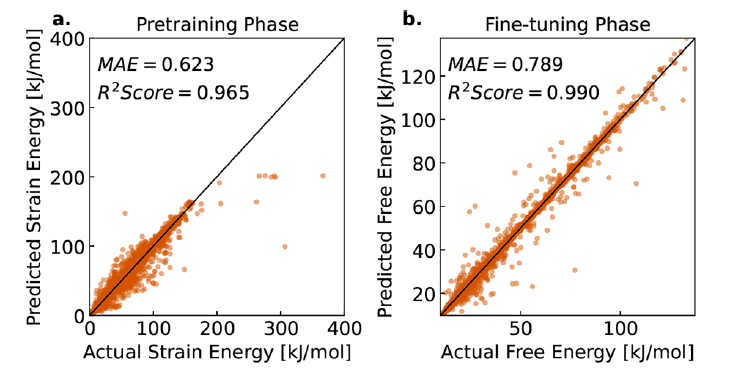
first,The team evaluated the free energy prediction performance of LLM-Prop on unknown MOF samples.The results show that the model can accurately predict the free energy with a mean absolute error (MAE) of 0.789 kJ/mol MOFatom, while achieving a high correlation of R² = 0.990, as shown in Figure b below.This means that the model can provide predictions that are close to the true values in the vast majority of MOF samples.
In the pre-training phase, the model was trained using strain energy data, achieving a MAE of 0.623 kJ/mol MOFatom and an R² of 0.965, as shown in Figure a. This high correlation indicates that strain energy data can provide effective preliminary information for free energy prediction, validating the rationality of the research team's pre-training strategy. Further analysis shows a high correlation between the pre-trained strain energy and the fine-tuned free energy, demonstrating the value of strain energy as a low-cost surrogate indicator in model training.
Ablation Experiment Results
To gain a deeper understanding of the sources of model performance, the team conducted systematic ablation experiments. The experiments examined the impact of local features, global features, and pre-training on free energy prediction. The results are shown in the table below:
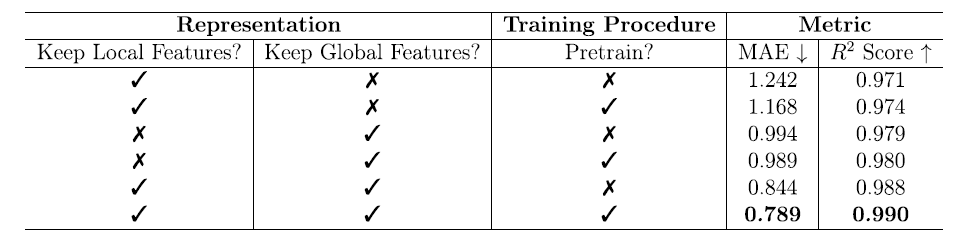
Local features only: Through pre-training, MAE decreased from 1.242 to 1.168 kJ/mol MOFatom, and R² increased from 0.971 to 0.974, indicating that pre-training can improve the model's generalization ability when local features are limited.
* Global features only:
The performance is significantly better than using only local features, with MAE decreasing to below 1.0 kJ/mol MOFatom and R² increasing to approximately 0.980. Pre-training has a relatively small impact in this case (MAE decreases from 0.994 to 0.989 kJ/mol MOFatom, and R² increases from 0.979 to 0.980), indicating that global features themselves contain more task information and require less reliance on pre-training to achieve effective learning.
* Combination of local and global features:
With the support of pre-training, the model achieved optimal performance with an MAE of 0.789 kJ/mol MOFatom and an R² of 0.990, demonstrating that the synergistic effect of the two types of features is crucial for improving prediction accuracy.
This ablation experiment clearly demonstrates that the design of global and local features and the pre-training strategy of MOFSeq are the core elements for improving the model's predictive ability.
Feasibility assessment of synthesis
In industrial applications, a more critical task is to determine the synthetic feasibility of MOFs, rather than simply focusing on the absolute value of the free energy. The research team set ΔL_MFFL (an index based on the free energy correction) to a threshold of 4.4 kJ/mol MOFatom and performed a binary classification prediction of the synthetic feasibility of MOFs. The experimental results are shown in the figure below:
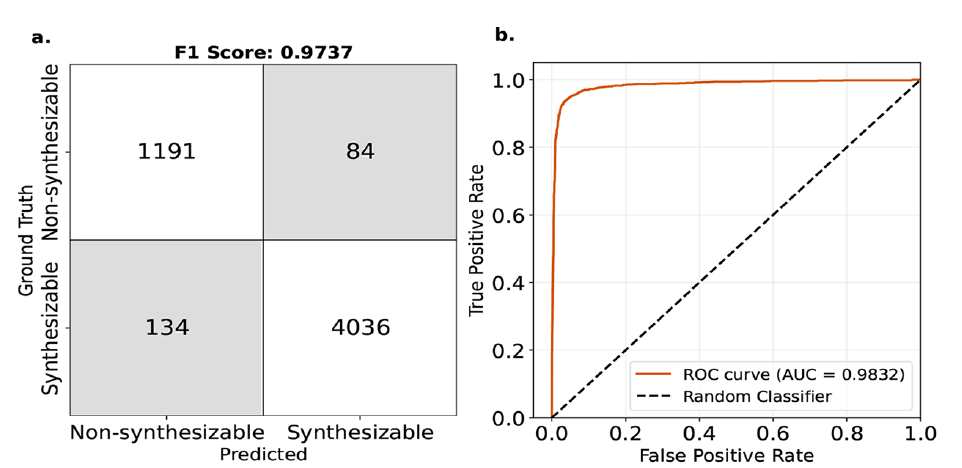
* The F1 score reached 97%—demonstrating the model's good generalization ability.
* The area under the ROC curve (AUC) is as high as 0.98 – which can ultimately be understood as the probability of an incorrect assessment if the model determines that a certain MOF is synthesizable, being only about 2%.
Screening of polymorphic MOFs
For MOF systems with polymorphism,The experiment further verified the model's ability to identify the most stable polymorph.Among 7,490 polymorph families, each containing 2–50 polymorphs, the model can correctly select the most stable polymorph with a free energy difference of only 0.16 kJ/mol MOFatom, with a success rate of approximately 63%; when the free energy difference increases to 0.49 kJ/mol MOFatom, the success rate increases to 89%.
Overall, the model achieved an average success rate of approximately 781 TP3T on the polymorphism recognition task.As shown in the figure below, it has significant value in high-throughput prediction before experimental screening.

From a practical application perspective, if LLM determines that a certain MOF design can be synthesized under the evaluation of thermodynamic stability and polymorphic competition, its correctness probability is between approximately 76% and 98%. The higher probability corresponds to the case where the MOF does not have competing polymorphs.
AI is reshaping the paradigm of MOF and materials science research.
October 8, 2025The Royal Swedish Academy of Sciences has decided to award the 2025 Nobel Prize in Chemistry to Professor Susumu Kitagawa of Kyoto University, Professor Richard Robson of the University of Melbourne, and Professor Omar Yaghi of the University of California, Berkeley, in recognition of their research contributions to the field of MOFs.Looking back from this historic moment, MOF research has traversed more than 30 years of development, gradually moving from initial structural construction and synthesis exploration to performance regulation, application expansion, and industrialization. Standing after this milestone, materials science is welcoming a new variable—the deep involvement of artificial intelligence is reshaping the research paradigm and innovation rhythm of MOFs and even the entire field of materials science.
In response to the challenge of a vast, complex world of MOFs lacking standardized naming conventions, in October 2025,A research team from the University of Toronto and the Clean Energy Innovation Research Centre of the National Research Council of Canada proposed MOF-ChemUnity: a structured, scalable, and extensible knowledge graph.This method utilizes LLM to establish a reliable one-to-one mapping between MOF names and their synonyms in the literature and crystal structures registered in CSD, thereby achieving disambiguation between MOF names and their synonyms and crystal structures. In the current version, MOF-ChemUnity integrates approximately 10,000 scientific articles and over 15,000 CSD crystal structures and their computational chemical properties, presented in a machine-operable format.
Paper title: MOF-ChemUnity: Literature-Informed Large Language Models for Metal–Organic Framework Research
Paper address:https://pubs.acs.org/doi/10.1021/jacs.5c11789
In the rational design of MOF materials, pre-synthesis structural prediction has always been a key challenge in achieving efficient and targeted synthesis of such materials. To address this,A team led by Professors Cui Yong and Gong Wei at Shanghai Jiao Tong University has developed a data-driven machine learning workflow that enables fast and accurate prediction of the metal node type of MOFs.This method uses the structural information of organic ligands as input and establishes a mapping relationship between ligand features and metal node types through a machine learning model, thereby effectively predicting the types of metal nodes that may be formed before synthesis. The trained and optimized machine learning prediction model achieved a prediction accuracy of 91%, a precision of 89%, and a recall of 85% on the test set.
Paper title: Data-Driven Machine Learning Assisted Prediction of Metal Node Types in Metal-Organic Frameworks for Guiding Linker Design and Targeting Inverse C3H8/ C3H6 Separation
Paper address:http://engine.scichina.com/doi/10.1007/s11426-025-2917-4
Traditional MOF research often starts with structure or properties, gradually approximating the target material through local variable control and extensive experiments or calculations. However, in these new works, the starting point itself is shifting—researchers are first constructing computationally achievable and reasonable material representation systems, and then allowing models to learn which structural combinations are physically plausible, thermodynamically feasible, and synthetically worthwhile. When models can quickly provide reliable thermodynamic and structural judgments in a million-scale structure space, the focus of materials research will shift upwards—from "how to calculate and measure" to "how to define the problem, construct representations, and set decision boundaries." This may be the next methodological leap that MOF research is poised to achieve after more than thirty years of structural and chemical accumulation.
References:
1.https://pubs.acs.org/doi/10.1021/jacs.5c13960
2.https://phys.org/news/2026-01-tool-narrows-ideal-metal-frameworks.html