Command Palette
Search for a command to run...
MIT Team open-sources BoltzGen, Enabling the Design of Protein Binders Across Molecular Types, Achieving Nanomolar Affinity for the 66% target.

In the fields of drug discovery and biomolecular engineering, de-novo binder design is a core method for automated drug discovery. Using computational simulation and deep learning, researchers can generate peptide or protein structures capable of binding to specific targets, enabling the development of novel drug modalities such as antibodies, nanobodies, and cyclic peptides.
However, traditional protein design strategies mostly rely on physical calculations such as molecular dynamics simulations and sequence optimization algorithms. Although high precision can be achieved in a single system,However, the computational cost is high, the design space is limited, and it is difficult to simultaneously process multimodal targets such as proteins, small molecules, and RNA.While current deep generative models have improved generation speed to a certain extent, they generally lack "atomic-level" structural reasoning capabilities and are optimized for specific categories of molecules, resulting in limited versatility. Furthermore, model evaluation often relies on existing similar complexes in the training set, making it difficult to verify generalization capabilities for "unseen targets." They lack controllable generation mechanisms and flexible structural constraint expressions, leading to limitations in design efficiency and interpretability.
To address this issue,MIT, in collaboration with Boltz and other institutions, has proposed the "All-atom Generative Model" BoltzGen, which unifies structure prediction and complex design.This model not only replaces traditional discrete residue labels with geometric continuous representations to achieve joint training of protein folding and binding design in a single system, but also constructs a flexible "design specification language" to achieve controllable generation across molecular types.
The experimental results show thatBoltzGen's nanobody and protein conjugate designs all have the goal of achieving nanomolar affinity for 66%.For the first time, it was demonstrated that a "single model system" can achieve simultaneous optimization of folding and binding performance in multimodal biomolecule design.
Currently, the relevant research results have been published under the title "BoltzGen: Toward Universal Binder Design".
GitHub address:
https://github.com/HannesStark/boltzgen
Research highlights:
* Unified structure prediction and binder design in a single all-atom generative model, enabling simultaneous protein folding, binding site modeling, and sequence generation at atomic-level precision, significantly improving the physical rationality and controllability of molecular design;
* A universal "design specification language" is proposed, allowing the model to flexibly switch between different systems such as proteins, nanoantibodies, cyclic peptides, and small molecules, realizing cross-modal structure generation and constraint control, and broadening the scope of application of generative AI in the field of biomolecular design.

Paper address:
https://go.hyper.ai/3sx2K
Follow the official account and reply "BoltzGen" to get the full PDF
More AI frontier papers:
Mixed datasets: multimodal training strategies
The research team adopted a multi-level, cross-modal joint training framework when training BoltzGen.The core sources of the datasets used include three categories:
* High-quality experimental structures from the Protein Data Bank (PDB), covering a variety of complex structures such as RNA, DNA, and protein small molecules, providing realistic chemical bond constraints and three-dimensional geometric distribution data for the model;
* Experimental data from the AlphaFold Database (AFDB), predicted and relearned by AlphaFold2, covering reliable folding patterns generated by experiments;
* The composite structure samples generated by the Boltz-1 model cover multimodal scenarios such as small molecule binding and RNA-DNA interactions, which can enhance the model's generalization ability across different biomolecule types.
To prevent the model from being overly biased towards specific structural types, the research team eliminated upsampled datasets for antibodies and TCRs to maintain diversity in the generated space. Furthermore, all structural samples were randomly cropped and multi-tasked during training, allowing the model to randomly handle tasks such as folding prediction, complex design, and structure completion in each training iteration. This unified, multifunctional learning framework enables the model to generate structures at the atomic level while also possessing cross-modal understanding capabilities.
Model Architecture: All-Atom Inference from Noise to Structure
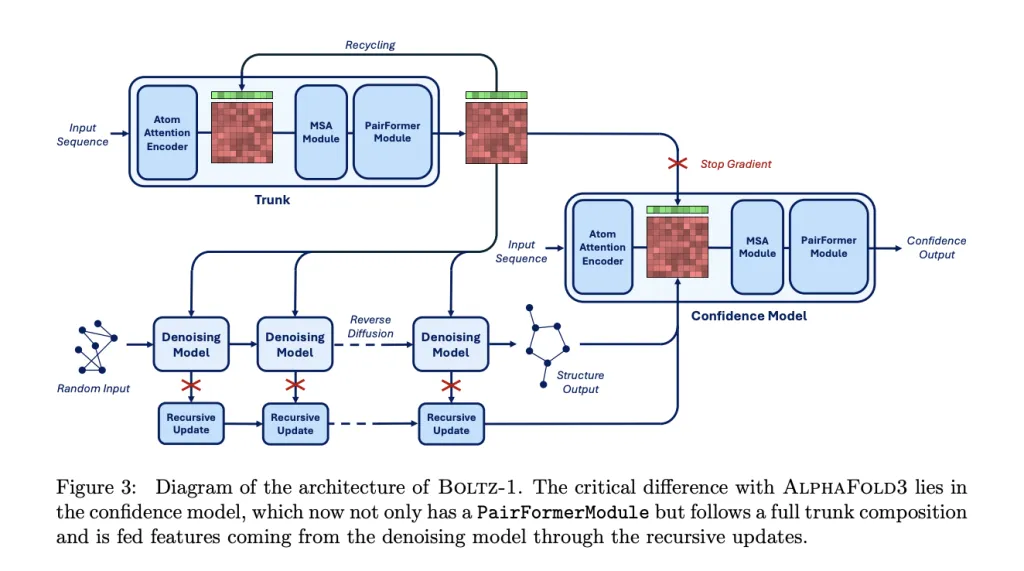
The model retains the main components of the AlphaFold3 and Boltz-2 architectures and makes some improvements on this basis to introduce more conditional inputs.
As shown in the figure below, the entire model is divided into two main parts:A larger Trunk (backbone network) and a Diffusion Module (diffusion module).The Trunk module generates token and pairwise representations for conditional control, while the Diffusion module generates the 3D structure based on these representations. The Trunk module runs only once, while the Diffusion module runs multiple iterations to gradually denoise the 3D coordinates of all atoms.
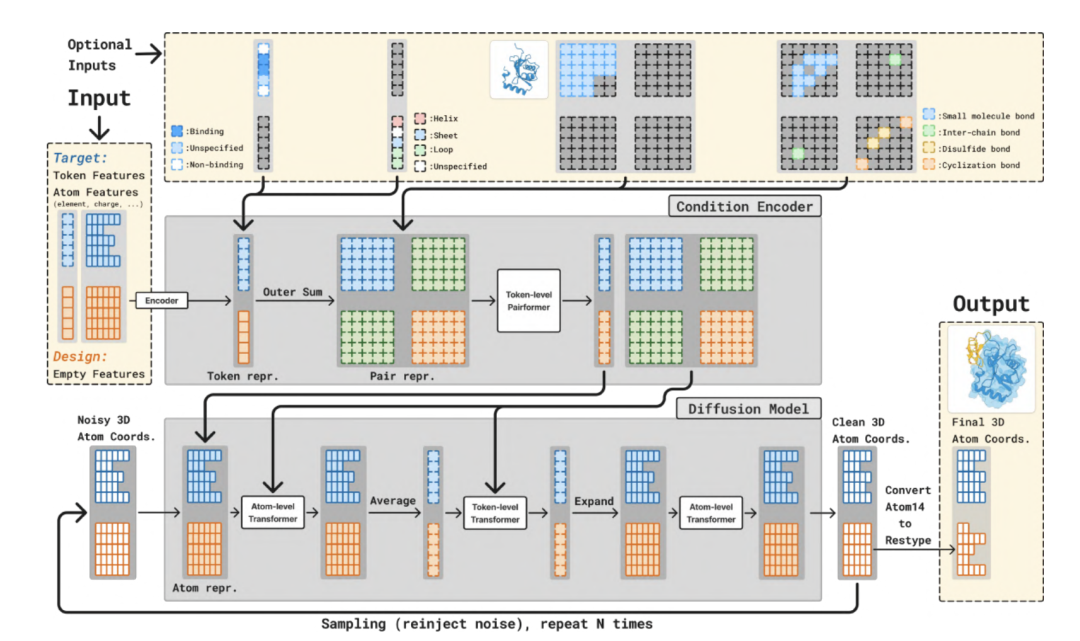
In the Trunk stage, it is similar to the Trunk module of Boltz-2 and is responsible for parsing the input protein structure and target information. The Trunk module processes tokenized molecular structures.The main framework uses a PairFormer architecture, utilizing Triangle Attention to efficiently model the spatial relationships between atoms. Combined with Geometric Residue Encoding, it simultaneously infers residue types and atomic coordinates in a continuous space, eliminating the reliance on discrete amino acid labels. This mechanism enables the model to truly understand the physical laws of the structure at generation time, rather than relying solely on data memorization.
In the Diffusion Module stage,This module receives noisy 3D atomic coordinates as input.and predicts its denoised coordinates. It employs a standard Transformer architecture, operating at both the atom and token levels. BoltzGen utilizes a continuous-space diffusion model to gradually "denoise" atomic coordinates. It predicts noise vectors to transform random initial states to stable conformations, preserving the constraints of the molecular energy surface during the generation process to avoid physical conflicts or structural collapse.
Experimental results: Universal design validation across 26 targets
In the experimental part, the performance verification of the BoltzGen model covered multiple dimensions from proteins to peptides, from novel pathogens to small molecule targets, demonstrating excellent generalization and controllability.
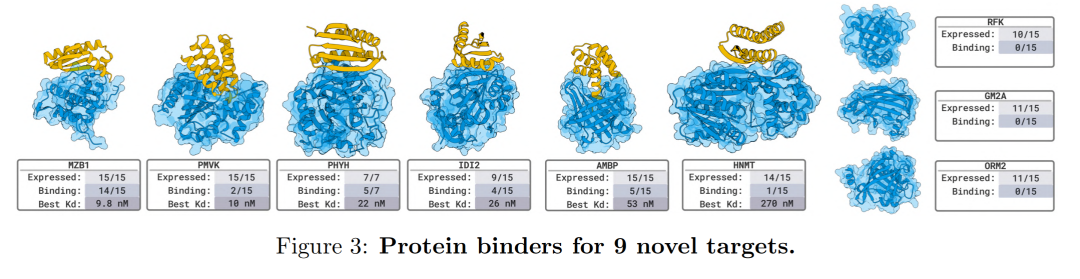
The team tested a total of 26 targets in 8 independent wet lab validation projects.The results involved a variety of binding types, including nanobodies, proteins, and linear and cyclic peptides. BoltzGen maintained a high success rate against unseen, complex targets: in nine experiments with novel targets completely different from the training data, the designed proteins and nanobodies all achieved nanomolar (nM) high-affinity binding to the 66% target, demonstrating the model's powerful structural reasoning and cross-modal design capabilities.
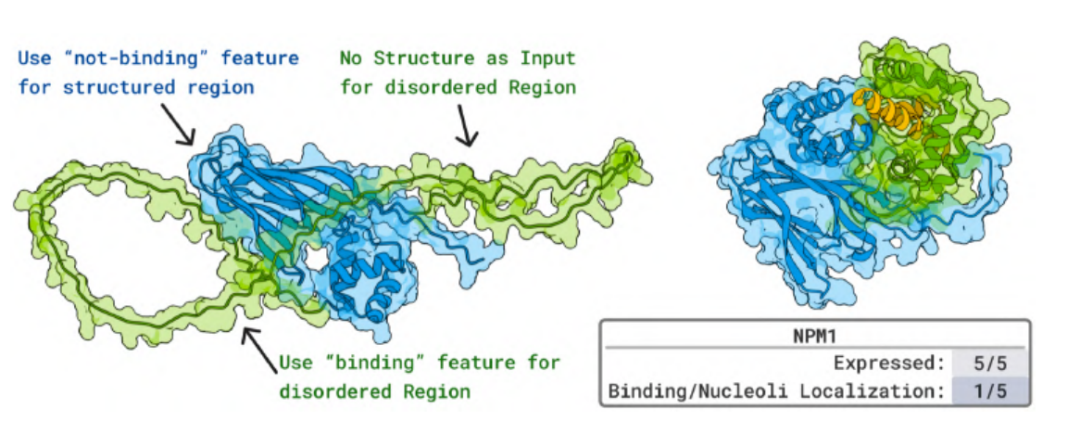
In experiments on bioactive peptides with diverse structures,BoltzGen-designed proteins can bind to different types of peptide molecules with nanomolar to micromolar (μM) affinities and effectively neutralize their antimicrobial or hemolytic activity. For the disordered protein NPM1, which is associated with acute myeloid leukemia, the peptides generated by the model exhibited nucleolar colocalization in living cells, providing the first in vivo evidence supporting the ability of AI-designed proteins to bind to naturally disordered proteins.
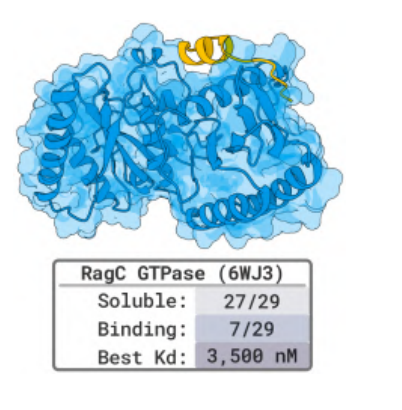
The design of RagC, a core enzyme in cellular metabolism, and RagA:RagC dimers also yielded remarkable results:Seven of the 29 candidate peptides successfully bound to RagC, with the highest affinity reaching 3.5 μM; 14 of the cyclic disulfide bond peptide designs showed stable binding.
BoltzGen also demonstrated cross-scale design capabilities on two small molecules of biomedical interest.The resulting protein binders displayed detectable binding activity in the 50–150 µM range, demonstrating that the model can achieve small molecule recognition without the need for expert chemistry guidance. Furthermore, in the design of antimicrobial peptides targeting the bacterial DNA gyrase GyrA, candidate sequences exceeding 19% reduced bacterial growth by more than fourfold, with some peptides directly killing host cells.
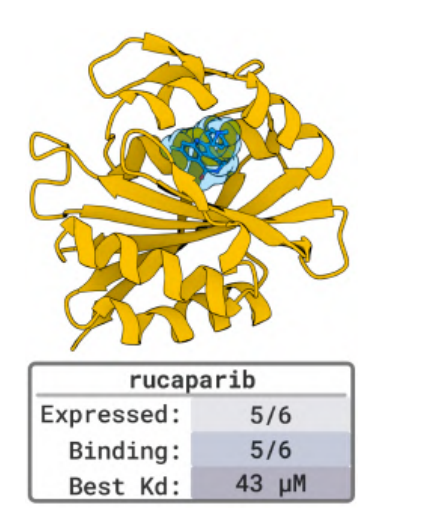
In the 5 benchmark target tests with known binding structures (such as PD-L1, TNFα, PDGFR, etc.),BoltzGen also achieved a high hit rate—nanomolar binders appeared on the target of 80%, verifying that its accuracy is on par with the current best model.

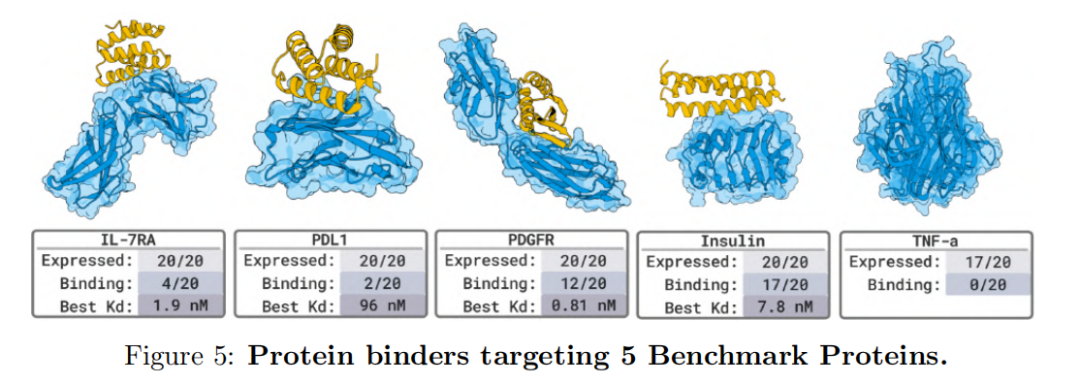
Overall, this series of experiments demonstrates that BoltzGen can not only reproduce high-quality binding structures within known data distributions but also achieve functional design in completely unfamiliar biological systems. Its unified all-atom generation architecture integrates the "design-prediction-verification" process, providing an open, controllable, and scalable AI infrastructure for future drug discovery and biomolecular engineering.
From prediction to generation, the Boltz series reshapes the landscape of AI-driven molecular design
In 2024,The MIT Jameel Clinic research team introduced the Boltz-1 model.As the global drug design industry shifts from structure prediction to function generation, while the AlphaFold series of models pioneered the computability of protein folding, the limited availability of AlphaFold3 restricts the industry's ability to freely iterate in real-world drug scenarios. Boltz-1 was born in this context. Not only does it approach AlphaFold3 in performance, but it is also fully open source and commercially viable, propelling molecular structure prediction into the industry's open ecosystem.
Boltz-1 uses a generation system that combines a diffusion model with a Transformer architecture.It can predict the structures of proteins, RNA, DNA, and small molecule complexes at the atomic level. Its flexible conditional interface enables precise modeling of specific binding sites or molecular conformations, significantly broadening its industrial application. From novel antibody design and enzyme engineering optimization to small molecule ligand screening, end-to-end predictions can be achieved within the Boltz-1 framework, significantly lowering the barrier to entry for biocomputing.
In 2025,The MIT Jameel Clinic team introduced the Boltz-2 model based on Boltz-1.It has pushed the accuracy of protein folding prediction to a new high and is known as the "GPT-4 of structural biology."
Compared to its predecessor, Boltz-2 achieves significant improvements in generation accuracy and computational efficiency. It also introduces multimodal conditional input, enabling it to integrate sequence information, experimental data, and chemical properties, enabling more refined molecular design. As the global biocomputing and drug discovery landscape shifts toward "full-scenario generation," Boltz-2 further addresses the demand in academia and industry for highly available, scalable, and commercially viable tools.
Boltz-2 inherits and optimizes the hybrid generation system of the diffusion model and the Transformer architecture.Its core Trunk module can extract multi-level representations of protein or nucleic acid complexes at one time.The Diffusion module generates and optimizes the structure based on this.
Thanks to a flexible conditional interface, researchers can precisely control the output structure for specific binding sites, active pockets, or small molecule ligands, significantly expanding the model's potential for application in areas such as novel antibody design, enzyme catalysis optimization, and drug lead screening. Boltz-2's open-source nature also ensures free iteration across academia and industry, accelerating the application of molecular generative computation in real-world drug development scenarios.
Today, BoltzGen has proposed a universal "design specification language" that allows the model to flexibly switch between different systems such as proteins, nanoantibodies, cyclic peptides, small molecules, etc., to achieve cross-modal structure generation and constraint control, further broadening the scope of application of generative AI in the field of biomolecular design.








