Command Palette
Search for a command to run...
Selected for NeurIPS 2025, the University of Toronto and Others Proposed a Ctrl-DNA Framework to Achieve "targeted Control" of Gene Expression in Specific cells.

Precise regulation of gene expression in specific cells is crucial for progress in fields such as gene therapy and synthetic biology. This process relies on a class of DNA sequences called “cis-regulatory elements (CRE)”, such as promoters and enhancers. They act like “switches” for genes, determining whether genes are “turned on” or “turned off” in target cells while avoiding abnormal activation in other normal cells. However,The number of naturally occurring effective CREs is limited and difficult to accurately match with diverse biomedical application scenarios.More importantly, the possibilities of DNA sequences are growing exponentially. For example, a 100-base sequence has 4¹⁰⁰ combinations. It is extremely difficult to verify them one by one through experiments. It is not only time-consuming and labor-intensive, but also cannot meet practical needs.
Current deep learning-based methods have greatly improved experimental efficiency, but existing methods still face multiple challenges.For example, some methods rely on mutations in existing DNA or random sequence optimization, which can easily fall into the trap of "local optimality," resulting in insufficient diversity in the generated effective sequences. While approaches based on autoregressive language models can capture DNA sequence patterns, they can only "mimic known sequences" and are unable to explore new cell-specific CREs. While reinforcement learning (RL)-based methods improve regulatory effects in target cells, they overlook the control of "side effects" on other cells. Furthermore, these standard design frameworks often overlook considerations of biological plausibility. The generated sequences may fail to match key transcription factor binding sites (TFBSs), leading to the failure of actual regulatory functions.
To fill the gap in the precise design of cell-specific CRE, a team from the University of Toronto, in collaboration with Changping Laboratory and other institutions, developed a constrained reinforcement learning framework called Ctrl-DNA.This framework, based on a pre-trained DNA language model, uses a reinforcement learning algorithm to simultaneously achieve dual objectives during the optimization process: maximizing the regulatory activity of CREs in target cells while strictly limiting their activity in non-target cells. Furthermore, the mathematical tool of Lagrange multipliers is employed to balance these two requirements, and the distribution of TFBSs in real DNA is referenced to ensure the biological validity of the generated sequences.
The results of the study showed thatIn the design tasks of 6 human cells, the CRE generated by Ctrl-DNA significantly outperformed the existing methods in two key indicators: "high activity in target cell types" and "constraint in non-target cell types".It also maintains significant diversity, providing new solutions for synthetic biology to "create controllable systems", gene therapy to "avoid off-target risks", and precision medicine to "perform cell-level customization".
The relevant research results were published on the arXiv preprint platform under the title "Ctrl-DNA: Constrained Reinforcement Learning for Cell-Specific Cis-Regulatory Element Design" and were selected for NeurIPS 2025.
Research highlights:
* A novel constraint-aware reinforcement learning framework is proposed to provide tools for designing CREs for precise cell-type-specific gene expression.
* Simplified the optimization process, improved experimental efficiency, and reduced computing costs
* Experiments have verified that Ctrl-DNA has both functional effectiveness and biological plausibility

Paper address:
https://arxiv.org/abs/2505.20578
Follow the official account and reply "Ctrl-DNA" to get the full PDF
More AI frontier papers:
Dataset: Based on real human promoter and enhancer datasets
In this study, researchers used real human promoter and enhancer datasets to evaluate and validate Ctrl-DNA.
in,The Human Promoter Dataset contains promoter activity data from three leukemia-derived cell lines.The three cell lines are: Jurkat, K562, and THP1. All three are mesoderm-derived hematopoietic cell lines with high biological similarity. Each sequence in this dataset is 250 base pairs long. See the table below:
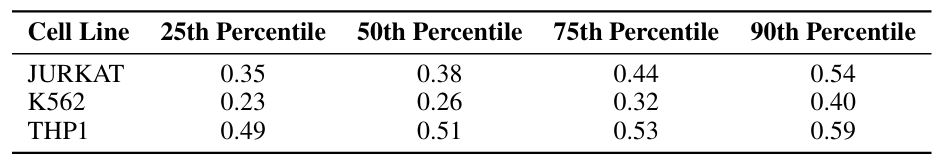
The Human Enhancer Dataset contains CRE activity data from three cell lines measured by a massively parallel reporter assay (MPRA).The three cell lines are: HepG2 (liver cell line), K562 (erythroid cell line), and SK-N-SH (neuroblastoma cell line). Each sequence in this dataset is 200 base pairs long. As shown in the following table:
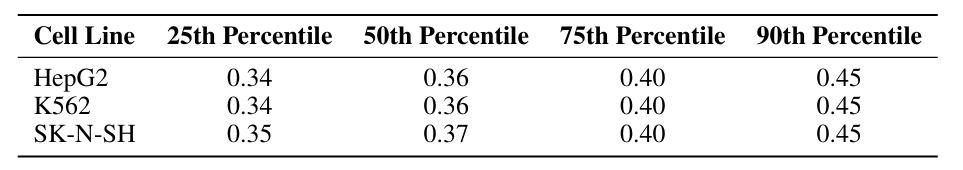
It is noteworthy that in the THP1 cell line, the 25th percentile activity reached 0.49, showing a right-skewed distribution. This distribution bias may partly account for the increased difficulty in constraining activity in the THP1 cell line.
Model architecture: Based on pre-trained DNA language model, combined with Lagrangian relaxation
Ctrl-DNA is a regulatory DNA sequence design framework based on constrained reinforcement learning, whose core goal is to generate CREs with controllable cell type specificity.In terms of functional implementation, it must maximize the fitness of the CRE in target cells—that is, enhance gene expression—while strictly controlling the fitness in off-target cells within a preset threshold. At the same time, it must ensure that the generated sequences conform to real biological laws to avoid situations where the experimental results meet the requirements but the application is ineffective.
To this end, researchers considered the framework's ease of use, rationality, and other aspects, and conducted a detailed design of the framework, as shown in the following figure:
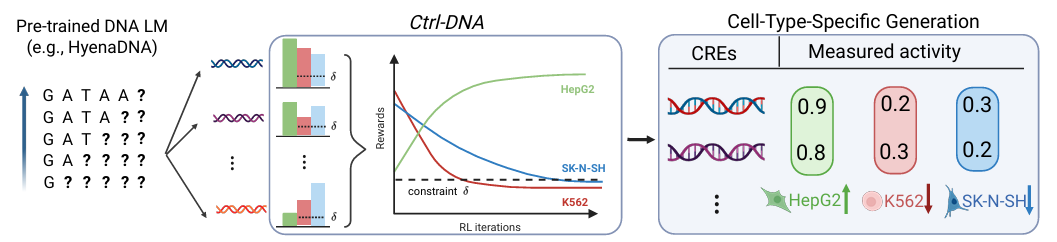
In terms of models and inputs,Ctrl-DNA fine-tunes the HyenaDNA autoregressive genomic language model pre-trained on the human genome as the initial policy model, and uses the Enformer architecture to train the cell type specific reward model.Combined with the "sequence-fitness" data measured by massively parallel reporting experiments, the target cell rewards and off-target cell rewards are calculated separately.
At the problem modeling level,The researchers transformed DNA sequence design into a constrained Markov decision process (CMDP). The core optimization mechanism of Ctrl-DNA utilizes Constrained Batch-wise Relative Policy Optimization (CBROP). This mechanism transforms a constrained optimization problem into an unconstrained primal-dual optimization problem through Lagrangian relaxation. The optimization process is iterative, with policy updates following the gradient of the Lagrangian objective function at the learning rate. Off-target cell rewards are constrained by adjusting the Lagrangian multiplier—increasing the Lagrangian multiplier to strengthen the constraint for off-target cells exceeding a threshold, and decreasing the Lagrangian multiplier to weaken the constraint for off-target cells meeting the threshold.
In order to reduce the complexity of training, Ctrl-DNA abandons the reliance on value models in traditional reinforcement learning.The normalized advantage is calculated directly based on batch data statistics to guide the strategy optimization to select sequences with "high target reward + low off-target reward".
In the design of the strategy update objective function, the researchers adopted a combination of "pruning replacement objectives" + "KL regularization". By pruning, they restricted strategy mutations and introduced the KL divergence between the current strategy and the initial reference strategy to ensure the consistency of the generated sequence with the natural DNA pattern, ultimately forming a strategy update objective function.
To further ensure biological plausibility, Ctrl-DNA introduces TFBS frequency correlation as an additional constraint. First, TFBSs are scanned using the FIMO tool from real, highly specific CRE sequences to construct a true TFBS frequency vector. The corresponding TFBS frequency vector is then calculated for each generated sequence. The Pearson correlation coefficient is then used as an additional constraint reward, while the corresponding Lagrange multiplier is clipped to [0, λmax] (λmax ≤ 1). This balances biological plausibility with objective optimization, avoiding over-constraints that could reduce model exploration capabilities.
To ensure model training stability, the researchers demonstrated the hyperparameter settings used in the experiments. All models were trained using the Adam optimizer, with a policy learning rate of 1e-4, a batch size of 256, and 100 training epochs. The experiments were trained on a single NVIDIA A100 GPU with 40GB of memory, as shown in the figure below.
Experimental results: Compared with 8 types of baseline methods, Ctrl-DNA has obvious advantages
The performance evaluation experiment of Ctrl-DNA revolves around two major design tasks: human enhancers and promoters, covering the six cell lines mentioned above. It is compared with eight types of baseline methods, including evolutionary algorithms (including AdaLead, Bayesian Optimization (BO), CMA-ES, PEX), generative models (RegLM), and reinforcement learning methods (including TACO, PPO, and PPO-Lagrangian), to verify its effectiveness and practicality from multiple dimensions such as cell type specificity, biological plausibility, and sequence diversity.
Ctrl-DNA exhibits significant advantages in terms of cell type-specific confinement.As shown in the figure below, the horizontal axis represents the fitness of off-target cell types, while the vertical axis represents the fitness of target cell types. The method shown in the upper right corner represents the best balance between maximizing target cell fitness and minimizing off-target expression.
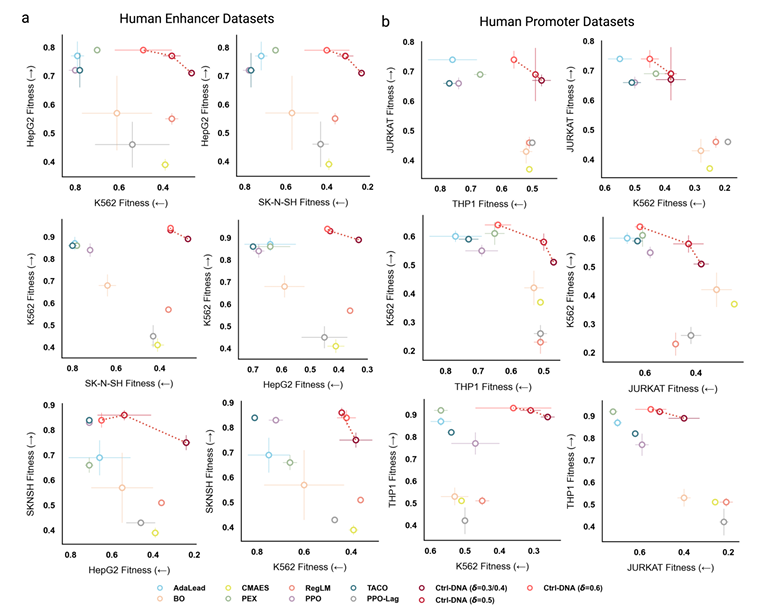
For enhancer design, Ctrl-DNA consistently achieved the highest target cell fitness while satisfying off-target constraints at all different constraint thresholds (δ = 0.3, 0.5, and 0.6). This means it maximized target cell fitness while strictly satisfying off-target constraints. Furthermore, while methods such as TACO and CMAES achieved high expression in target cells, they were unable to suppress off-target cell fitness, resulting in poor cell-type specificity.
For the promoter design task, because all three target cell types are hematopoietic cells of mesoderm origin, they have great transcriptional similarities, which poses a significant challenge for this task, but Ctrl-DNA still performs well. The experiment set three different constraint thresholds (δ=0.4, 0.5, and 0.6) for testing. Ctrl-DNA outperformed all baselines when maximizing the fitness of the target cell type and meeting the constraint thresholds δ=0.5 and 0.6. It is also worth noting that for cases such as THP1 cells where the activity distribution is right-skewed (as mentioned in the dataset section above, the 25th percentile activity has reached 0.49), no method can suppress its off-target activity to the strict threshold of δ=0.4, but Ctrl-DNA is the method that comes closest to the constraint requirement among all methods.
In biological plausibility validation, as shown in the figure below, Ctrl-DNA achieved the highest reward difference (ΔR) across all cell types for human promoters and enhancers, indicating that it better optimizes the cell-specific fitness of DNA sequences. Regarding motif relevance, Ctrl-DNA also achieved stronger performance in most cell types, with the exception of the THP1 promoter design.
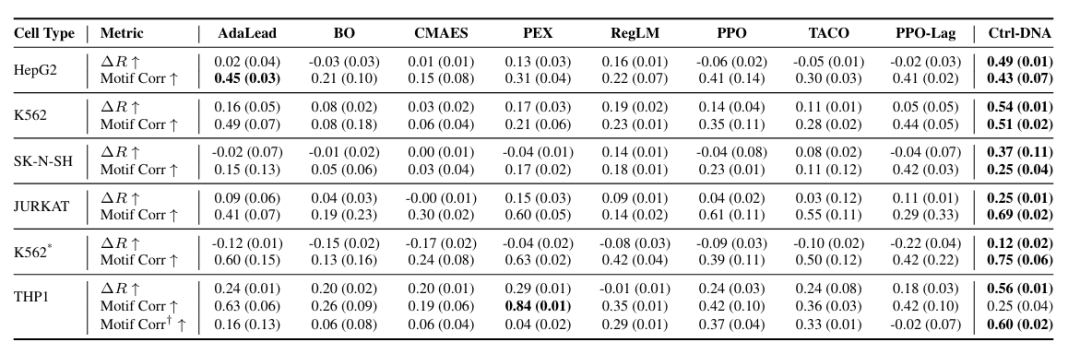
To further explore this discrepancy, the researchers extracted motifs from promoter sequences at the 90th percentile of THP1 fitness. Using a threshold of q < 0.05 to avoid false positives, they re-evaluated the motif correlation between the generated sequences and the reference set, represented as motif Corr† in the figure above. The results showed that Ctrl-DNA outperformed all baselines even under this stringent setting, with its correlation coefficient increasing to 0.60, while the correlations for most baselines decreased, demonstrating its ability to preferentially capture functionally significant regulatory motifs.
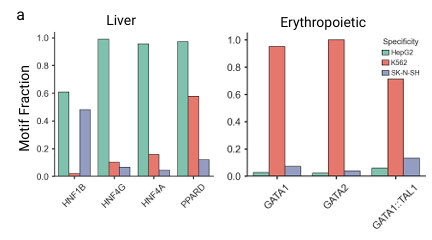
To further analyze the frequency of specific TFBSs found, the researchers specifically examined the sequences generated for motifs specific to the HepG2 hepatocyte cell line and the K562 erythroid cell line.As shown in the figure below, the HepG2 sequence generated by Ctrl-DNA displays the highest frequency of liver-specific motifs, such as HNF4A and HNF4G. Similarly, the sequence generated for K562 contains the highest frequency of erythroid-specific motifs, such as GATA1 and GATA2. This demonstrates that Ctrl-DNA not only optimizes the fitness of the target cell but also learns regulatory patterns that reflect underlying cell-type specificity.
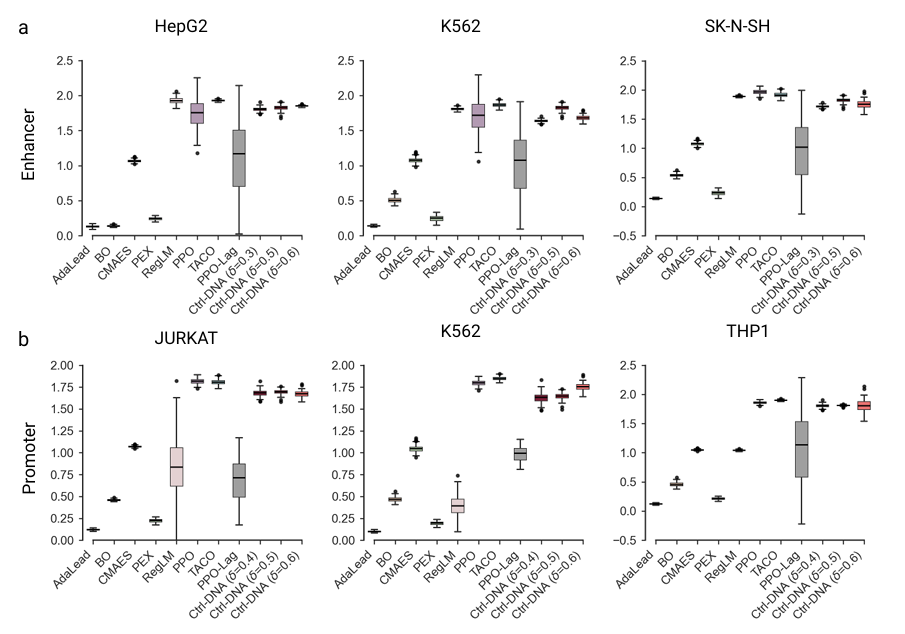
In terms of sequence diversity, Ctrl-DNA achieved comparable or higher diversity than most baselines, confirming its ability to generate diverse sequences without sacrificing regulatory control.As shown in the following figure:
Finally, the researchers further validated the effectiveness of the Ctrl-DNA core module through ablation experiments. The role of the TFBS regularization module was also confirmed, effectively guiding sequences toward biologically realistic patterns.
AI-driven DNA "switch" design opens a new chapter
In the past, the design of regulatory DNA sequence "switches" mostly relied on "trial and error" through a large number of repeated manual screenings.Now, with the combination of AI technology, we can use algorithms to predict "which DNA sequences have the highest match with the target regulatory protein", which greatly improves the design efficiency and accuracy.This is also the core reason why AI-driven DNA switch design has become a new direction, which in turn directly promotes fields such as gene therapy and synthetic biology from "extensive" to "precise".
This paper is just one of the big fruits on the big tree of "AI-driven DNA switch design". Looking back in the past, many laboratories have already carried out related research.
For example, a team from The Jackson Laboratory, the Broad Institute, and Yale University published a study in Nature titled "Machine-guided design of cell-type-targeting cis-regulatory elements."The study used artificial intelligence to design thousands of new DNA switches.These switches can precisely control gene expression in different cell types. Specifically, the researchers constructed a deep convolutional neural network (Malinois) that can accurately predict CRE activity and developed a modular platform (CODA) for designing CREs with specific functions. This platform provides powerful tools for developing reporter genes, CRISPR therapy, gene replacement methods, and more.
Paper address:
https://www.nature.com/articles/s41586-024-08070-z
In addition, there is RegLM mentioned in the above article, which comes from Genentec. In the study titled "Designing realistic regulatory DNA with autoregressive language models",We introduce a framework called RegLM, which is based on an autoregressive language model combined with a supervised sequence-function model to design synthetic CREs with specific properties.Similarly, RegLM is also based on the HyenaDNA framework. It encodes functional labels as hint tokens and adds them to the DNA sequence prefix, trains or fine-tunes the model to perform next token prediction, and thus generates DNA sequences with desired functions. At the same time, it combines a supervised sequence-activity regression model to screen the generated sequences.
Paper address:
https://genome.cshlp.org/content/34/9/1411.full#aff-1
In summary, the development of Ctrl-DNA is undoubtedly another step forward in DNA switch design. Although there are still some problems or areas that need urgent improvement, such as incorporating additional biological constraints to further improve the rationality and functionality of the generated sequence, and adjusting the Lagrange multiplier is still a matter of experience, the development and improvement of these tools have undoubtedly opened a new chapter for DNA switch design, while also promoting the continuous development of the interdisciplinary science of artificial intelligence and biology.








