Command Palette
Search for a command to run...
Solving the Atomic-Level Modeling Challenge of Protein Conformation Heterogeneity! David Baker's Team's PLACER Framework Analysis

In the molecular world, the interactions between proteins and nucleic acids, small organic and inorganic molecules, and metal ions are crucial to life functions. Every recognition and binding of these interactions can affect biological functions, determine drug efficacy, and even influence the success or failure of novel enzyme design. However, atomic-level modeling of these interactions and their conformational heterogeneity remains an extremely challenging task for the industry.
Deep learning (DL) based small molecule docking tools such as DiffDock have improved accuracy compared to earlier methods.However, the performance difference is not significant in high-precision tasks.Furthermore, performance significantly degrades when encountering unseen receptors; in addition, various deep learning-based methods have been developed to generate small molecule conformations from chemical structures.However, these methods typically model only specific categories of interacting objects, thus limiting their ability to characterize the full spectrum of protein function.
Based on this, a research team led by Nobel laureate Professor David Baker from the University of Washington developed a graph neural network called PLACER (Protein-Ligand Atomistic Conformational Ensemble Resolver).It can accurately generate the structures of various organic small molecules based on the atomic composition and bonding information of small molecules; and, given the macroscopic structural environment of proteins, it can construct the detailed structures of small molecules and protein side chains for protein-small molecule docking tasks.In enzyme design research, this research team discovered that using PLACER to assess the accuracy of designed active sites and the degree of pre-organization can significantly improve the design success rate and enzyme activity. For example, the researchers obtained a pre-organized antialdolase with kcat/KM = 11,000 M⁻¹·min⁻¹.It far surpasses all design results before the advent of deep learning.
The related research findings, titled "Modeling protein-small molecule conformational ensembles with PLACER," have been published in the Proceedings of the National Academy of Sciences (PNAS).
Research highlights:
* PLACER is high-speed and random, enabling it to quickly generate a large number of prediction samples to depict the distribution of conformational heterogeneity.
* By using a unified atomic-level representation for all interactions, PLACER can be easily extended beyond biomolecules, such as macrocyclic molecules and other complex small molecules.
* PLACER is of great value for computational enzyme design and small molecule conjugate design: it can rapidly assess the accuracy of the reconstruction of the designed active site and the pre-organization of key catalytic/interacting side chain functional groups.

Paper address:
https://www.biorxiv.org/content/10.1101/2024.09.25.614868v2
Follow our official WeChat account and reply "enzyme design" in the background to get the complete PDF.
Datasets: Multi-level and diverse data construction validates excellent generalization ability
For small molecule conformation prediction, the team selected over 226,000 organic non-polymer small molecule crystal structures from the Cambridge Structural Database (CSD) as the training set and chose 7,116 samples as the validation set. Each molecule provides complete atomic composition and chemical bond information, while atomic coordinates are randomly initialized, enabling the model to learn the ability to recover accurate structures under noisy conditions.This training strategy not only ensures that the model can capture the subtle changes of small molecules in different conformations, but also allows for the generation of a diverse set of molecular conformations through multiple runs.
Regarding protein-small molecule systems, the research team selected high-resolution (<2.5 Å) structures from the Protein Data Bank (PDB), including protein-small molecule complexes, totaling approximately 113,000 training samples and 7,090 validation samples. Notably,The team excluded only water molecules but retained information on potentially non-biological small molecules (such as solvents) because they still provided valuable clues about the physicochemical preferences of molecular interfaces.The training data is cropped to contain a maximum of 600 heavy atoms and perturbed with Gaussian noise around randomly selected atom centers to simulate the complex dynamic environment of proteins and small molecules in reality.
This multi-layered and diverse data construction ensures that PLACER exhibits excellent generalization ability when processing everything from single small molecules to complex protein-small molecule systems.
The neural network PLACER employs a three-track architecture, focusing on atomic-level side chains and small molecule conformations.
PLACER is a denoising neural network whose input includes partially perturbed protein structures and chemical structure information (excluding coordinates) of any interacting molecules. Its output is the full atomic structure of the complex and the uncertainty of each atom position in the prediction model, as shown in Figure A below.
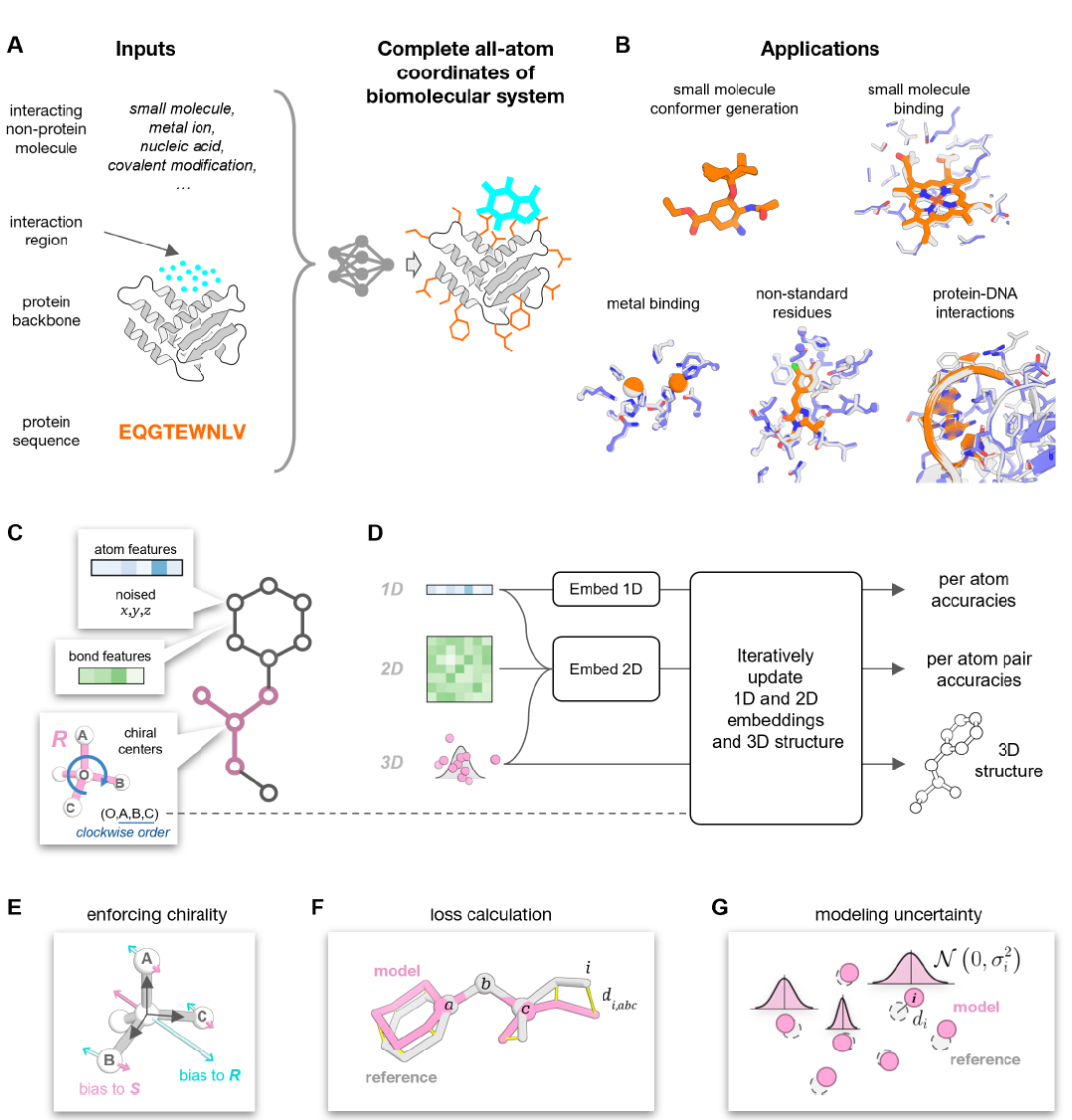
During the input phase, the molecular system is transformed into a chemical graph, where nodes represent individual heavy atoms (hydrogen atoms are not modeled to reduce computational costs), and edges represent chemical bonds between atoms (see Figure C above). This representation is consistent across different types of molecules. Each node in the network contains atom type information and its initially perturbed 3D coordinates. The network's task is to iteratively denoise the input coordinates while simultaneously estimating the uncertainties in the atomic positions within the output model structure (see Figure D above).
PLACER adopts a three-track architecture inspired by RoseTTAFold (RF), and the overall network architecture is as follows:
* Three-orbital design (1D, 2D, 3D): 1D orbitals handle atomic feature information; 2D orbitals handle inter-atomic pair relationships (such as chemical bonds and spatial proximity); 3D orbitals are responsible for updating atomic coordinates.
* Iterative Optimization: After the initial embedding of 1D and 2D features is completed, these features are passed to the iteration block to iteratively update the embedding vectors and 3D structure. In the iteration block, an atomic neighbor graph is first constructed—for each atom, half of the spatial proximity and half of the chemical graph proximity are selected, for a total of 32 nearest neighbor atoms. Then, the 2D pairs of features are projected into edge embeddings through a feed-forward adapter layer, and together with the 1D features, the atomic neighbor graph, and the current 3D atomic structure, they are used as input to the SE3-Transformer network to update the 3D coordinates and 1D embedding vectors.
* Chiral center processing: Chiral center information is passed to the network through Type-1 (vector) features (see Figure E above); features in 2D orbitals undergo pair-to-pair updates and are combined with structural biases. The confidence prediction heads for atoms and their pairs branch out from the 1D and 2D orbitals respectively to complete the computation of the iterative blocks; the fully trained network contains eight iterative blocks with shared weights.
* Loss Function Design: PLACER training uses a combination of structural loss and confidence prediction loss, applied after each iteration. The primary structural loss is all-atom FAPE (Frame-Aligned Point Error); the confidence of the model structure is evaluated at both the atomic and atom-pair levels.
Through this carefully designed network architecturePLACER can generate diverse and atomically precise sets of protein-small molecule conformations starting from randomly initialized coordinates.This provides a reliable foundation for subsequent analysis and design. Unlike protein structure prediction methods such as AlphaFold,PLACER does not predict the main chain structure of proteins, but focuses on atomic-level side chains and small molecule conformations, thus significantly improving computational speed and allowing the generation of diverse conformation sets.
Results Showcase: Providing Support for Precision Engineering from Small Molecules to Complex Protein Systems
Small molecule conformation prediction
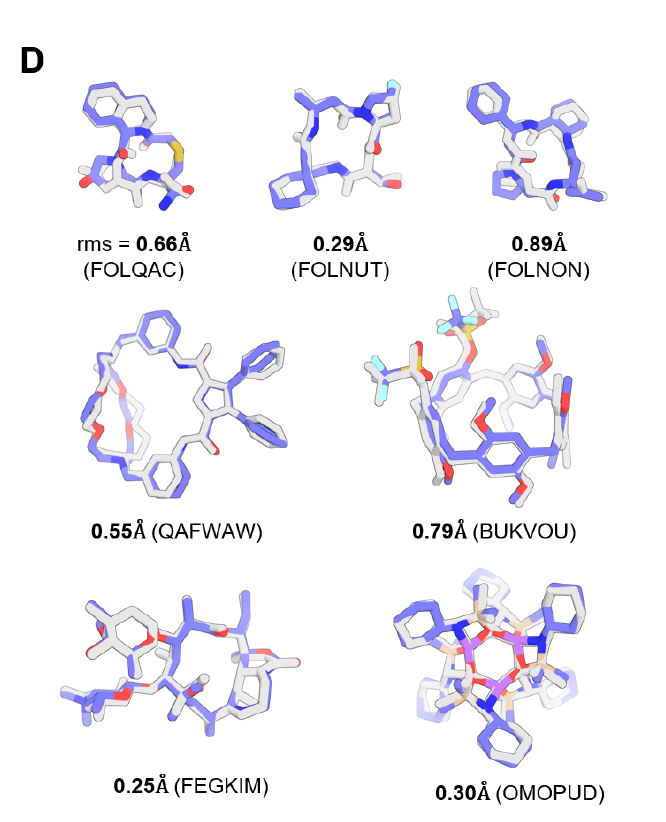
Tests on the CSD small molecule dataset demonstrate that the fully trained PLACER can correctly generate the three-dimensional structures of complex molecules with sub-Å accuracy.Examples include macrocycles with more than 50 atoms (see Figure D below), including peptide macrocycles (see the top row of Figure D below). Ablation experiments show that missing bond distance information or reducing the number of iterations significantly reduces prediction accuracy, demonstrating the critical importance of the iteration and feature strategies designed for PLACER.
Protein-small molecule interactions
Researchers used PLACER to generate a set of small molecule conformations in the pocket of the target protein (Figure A below) by running the network multiple times, each time with different random initializations of the input coordinates.Analysis of the generated conformation set shows that PLACER is insensitive to the initial position of the ligand: multiple different starting positions can produce predictions that are close to the natural conformation (Figure B below), and these positions cover the entire space of the input sampling (Figure D below).The researchers also observed that the predicted RMSD score (pRMSD) calculated based on ligand atoms could be used to select more accurate models from the sampling pool (Figure E below), with the highest-scoring model showing a high degree of agreement with the experimental structure (Figure C below).
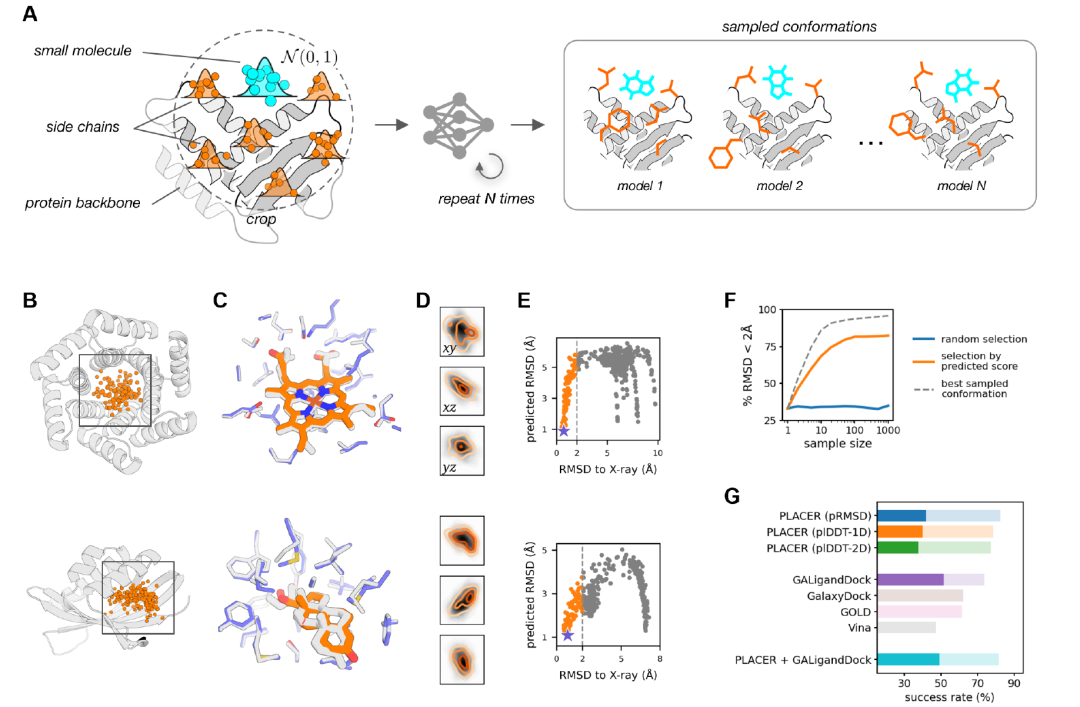
As shown in Figure G above, in the non-natural conformation test of 65 drug targets, PLACER performed excellently in generating and selecting near-native conformations, with a success rate using pRMSD scoring exceeding that of traditional docking tools such as Vina, GOLD, and GalaxyDock. Compared to the best-performing Rosetta GALigandDock method, PLACER performed better in the low-precision range (percentage of complexes with ligand RMSD < 2 Å) (82.4% vs 73.6%), but slightly worse in the high-precision range (RMSD < 1 Å) (41.8% vs 51.6%).
However, PLACER's performance is still noteworthy because, unlike other methods, it is not specifically trained for non-natural protein-small molecule docking tasks. PLACER can reconstruct the conformation of small molecules and protein side chains from scratch, while other testing methods mainly rely on the coordinates of the input protein.
Enzyme active site design
PLACER's application in retro-aldolase design is particularly noteworthy. The research team conducted 50 repeated simulations of the RA95 series of retro-aldolases and their evolved improved versions, analyzing the conformational diversity of the active site lysine and its covalent intermediate. The results showed that for enzymes with low activity in the initial computational design, PLACER generated highly diverse conformational sets, indicating a lack of pre-organization; while for the more active evolved versions, the conformational sets became increasingly ordered. This suggests that...The lack of pre-organization is a major shortcoming in early enzyme design, while PLACER provides a rapid evaluation tool that can be used to guide enzyme design work.
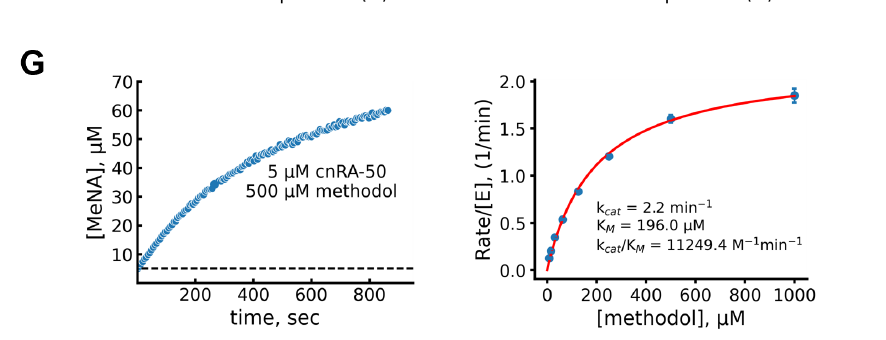
Furthermore, the team designed a new type of aldosterone reversal enzyme based on NTF2-like folding and assessed the correlation between pre-organization degree and kcat/KM value using PLACER. The results showed that PLACER predicted highly pre-organized designs generally had higher catalytic efficiency, with the most active design, cnRA-50, achieving a kcat/KM of 11,000 M⁻¹min⁻¹, significantly higher than earlier computational designs and approaching the activity of designs created using the latest RFdiffusion and proteinMPNN methods.
The research team predicts that,PLACER-based conformational set generation methods will be widely used for structural modeling of complex non-protein molecules in isolated states or protein environments, as well as for evaluating enzyme design and protein-small molecule conjugate design.
Professor David Baker: A pioneer who has long focused on computational protein design
On October 9, 2024, renowned protein design pioneer Professor David Baker, along with AlphaFold2 developers Demis Hassabis and John M. Jumper of DeepMind, were awarded the 2024 Nobel Prize in Chemistry.
Professor David Baker has long focused on computational protein design, open-sourcing deep learning tools such as RoseTTAFold, RFdiffusion, and ProteinMPNN to empower the design of novel proteins. He has also driven the industrialization of these technologies by founding a company, making him a true world-class master in the field. In his latest research, his team has achieved substantial breakthroughs in many new directions.
For example, when developing new drugs, researchers often use proteins as core drug targets, binding the drug to structurally stable proteins to intervene in disease progression. However, targeting naturally occurring disordered proteins (IDPs), which lack well-defined structure, sequence, and conformational preferences, remains challenging. Against this backdrop, in August 2025, David Baker's team proposed a protein design strategy called Logos, based on an induced fit binding strategy, designing binding proteins capable of adapting to 39 target disordered amino acid sequences. This means that more proteins can provide targets for new drug development, potentially accelerating research in cancer and Alzheimer's disease.
Paper Title:Design of intrinsically disordered region binding proteins
Paper address:https://www.science.org/doi/10.1126/science.adr8063
On September 18, 2025, David Baker's team proposed an all-atom diffusion model—RFdiffusion3 (RFD3)—that enables de novo design of all-atom biomolecular interactions. This model can generate protein structures in the context of ligands, nucleic acids, and other non-protein clusters, and is simpler and more efficient than previous methods. In a series of computer simulation benchmarks, RFdiffusion3 outperformed previous methods, with computational costs only one-tenth that of its predecessors.
Paper Title:De novo Design of All-atom Biomolecular Interactions with RFdiffusion3
Paper address:https://www.biorxiv.org/content/10.1101/2025.09.18.676967v1
Natural ion channels play a crucial role in biological systems, and their artificially engineered versions have been widely used in chemogenetic tools and sensors. While protein design has been used to construct transmembrane proteins with porous structures, designing "selective filters"—those with precise amino acid side chains that target specific ions—like natural ion channels has been a technological limitation. In October 2025, David Baker's team's latest research, for the first time, used artificial intelligence to design a novel calcium ion channel from scratch. This study demonstrates that even complex biochemical functions that are only partially understood by us can now be constructed from first principles using AI.
Paper Title:Bottom-up design of Ca² ⁺ channels from defined selectivity filter geometry
Paper address:https://www.nature.com/articles/s41586-025-09646-z
Looking at their recent achievements, Professor David Baker and his team are reshaping the landscape of protein science at an astonishing pace—from the Logos strategy, which can bind to naturally disordered proteins, to RFdiffusion3, which enables atomic-level molecular interaction design, and the groundbreaking research on the first de novo construction of calcium ion channels. Baker's team is continuously pushing computational protein design from theory to reality. Their work not only expands the boundaries of biomolecular design but also makes the future of "building life functions with algorithms" increasingly clear.
Reference Links:
1.https://www.biorxiv.org/content/10.1101/2024.09.25.614868v2
2.https://www.thepaper.cn/newsDetail_forward_31663354
3.https://www.biorxiv.org/content/10.1101/2025.09.18.676967v1
4.https://www.nature.com/articles/s41586-025-09646-z








