Command Palette
Search for a command to run...
Columbia University and Stanford University Collaborate! Squidiff Enables multi-scenario Transcriptome Simulation, Contributing to the Development of Precision Medicine and Space medicine.
In cell biology research, living cells are always complex dissipative systems far from chemical equilibrium, and how their collective response to external stimuli remains a core scientific question that scientists strive to uncover. This response is not only jointly regulated by internal tissue heterogeneity and external signals, but also often exhibits unpredictable nonlinear dynamic characteristics. Although single-cell sequencing technology allows us to unbiasedly analyze the heterogeneous composition of cells, accurately tracing the changes in the entire transcriptome after stimulation still faces significant challenges.
To overcome this limitation, the scientific community has previously developed various machine learning models such as scGen and CellOT. However, these models perform poorly in predicting high-resolution dynamic transitions, and most models rely on task-specific design, significantly limiting their applicability. The advent of diffusion models has brought a new breakthrough to this field: by iteratively generating optimized data, richer data distribution characteristics can be captured, providing a new approach to solving the aforementioned problems. Currently, some studies have attempted to combine diffusion models with variational autoencoders (VAEs) or implement the diffusion process in the latent space, successfully generating high-fidelity single-cell data and improving modeling efficiency.However, the application of diffusion models in key scenarios such as gene perturbation response prediction, drug perturbation response prediction, and cell development trajectory inference remains an underdeveloped area..
In this context,Research teams from Columbia University, Stanford University, and others have developed the Squidiff computational framework.This framework is built on a conditionally denoised diffusion implicit model and can predict transcriptomic responses of different cell types under differentiation induction, gene perturbation and drug treatment.Its core advantage lies in its ability to integrate definitive information from gene-editing tools and drug compounds:In predicting stem cell differentiation, Squidiff can not only accurately capture transient cell states but also identify non-additive gene perturbation effects and cell-specific response characteristics. The research team further applied Squidiff to vascular organoid research, successfully predicting the effects of radiation exposure on various cell types and evaluating the protective efficacy of radiation-protective drugs.
The related research findings, titled "Squidiff: predicting cellular development and responses to perturbations using a diffusion model," have been published in Nature Methods.

Paper address:
https://www.nature.com/articles/s41592-025-02877-y
Follow our official WeChat account and reply "Squidiff" in the background to get the full PDF.
More AI frontier papers:
https://hyper.ai/papers
Dataset: Full coverage of multiple scenarios + standardized quality control
To fully train and validate the performance of the Squidiff framework,The research team constructed a multi-scenario dataset that includes both simulated and real experimental data, covering key research directions such as cell differentiation, gene perturbation, drug treatment, and radiation response of vascular organoids.All data underwent a unified quality control process: low-quality cells with a mitochondrial gene ratio exceeding 20% or fewer than 1,000 genes were filtered out, low-expression genes were removed, and in some scenarios, dual-cell and stress-related genes were further excluded. Finally, sequencing depth differences were corrected using log normalization to ensure comparability across datasets.
Regarding the simulated data, the team used the Splatter tool based on hierarchical gamma-Poisson distribution to generate synthetic single-cell RNA sequencing data, simulating the expression heterogeneity and variance characteristics of real scRNA-seq, to verify the model's basic capabilities in transcriptome reconstruction and inference without the need for additional biological preprocessing.
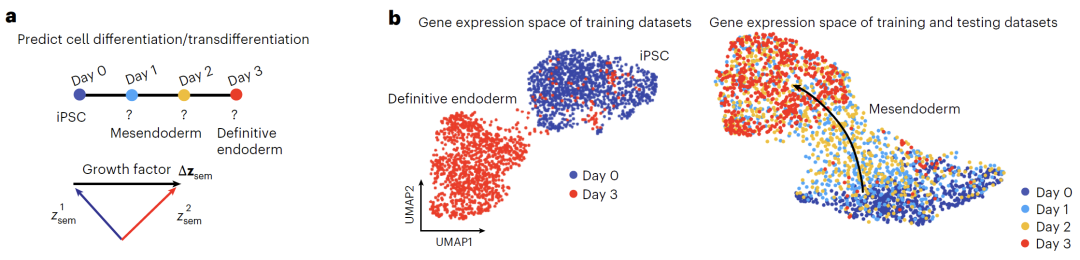
Cell differentiation data were derived from a publicly available dataset of human induced pluripotent stem cell (iPSC) differentiation into the endoderm, containing transcriptomes of 4,800 cells from day 0 (iPSC state) to day 3 (defined endoderm state). The model used day 0 and day 3 data as the training set and day 1 and day 2 data as the test set. The top 203 hypervariable genes were selected for modeling. Gaussian noise was introduced during training, and 1,000 diffusion steps were set. Differentiation semantic variables were obtained by calculating the average difference in latent representations, and then linear interpolation was used to simulate the developmental trajectory from day 0 to day 3 to evaluate the model's predictive ability for the dynamic differentiation process.
The gene perturbation data came from a CRISPR screening experiment on K562 cells.The study included approximately 10,000 cells, encompassing both ZBTB25 and PTPN12 gene knockout and their wild-type control. Data was divided into three groups: "PTPN12 + control," "ZBTB25 + control," and "PTPN12 + ZBTB25." The first two groups were used for training, and the last group was used for testing. After training, gene perturbation-specific variables were extracted and combined to simulate transcriptomic changes resulting from combined dual-gene perturbation, validating the model's ability to capture non-additive effects.
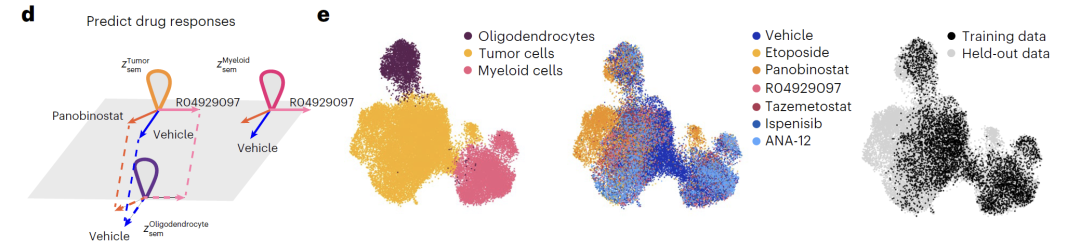
Drug processing data integrates multiple cell and drug samples.This includes expression profiles of glioblastoma treated with six drugs, including etoposide, and response data of melanoma to drug combinations. During training, the model learns specific perturbation representations for each drug and incorporates unknown drug samples from the sci-Plex3 dataset. By combining SMILES structure, dosage information, and compound fingerprints, it achieves generalized prediction of the perturbation effects of unknown drugs.
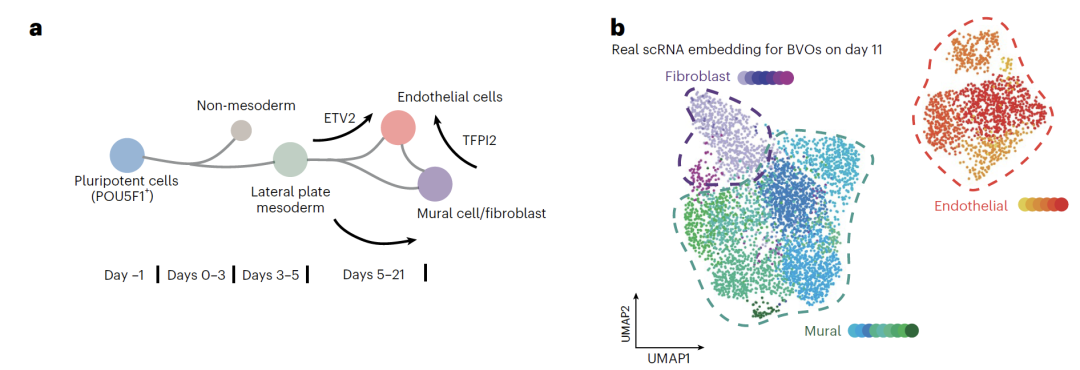
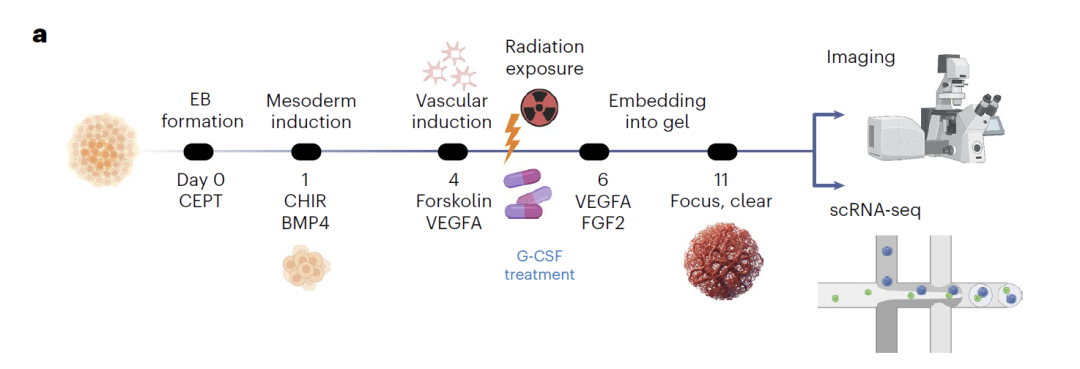
The vascular organoid data is based on original experiments.Endothelial cells, parietal cells, and fibroblasts were differentiated from healthy human iPSCs. On day 5, the cells were subjected to neutron or photon radiation, and scRNA-seq data were collected on day 11, forming a resource library encompassing 72 organoids and approximately 60,000 cells. Multimodal validation was further provided by ELISA measurements of inflammatory factors. In modeling, the team used data from day 0 and day 11 to train the model and interpolate to predict cell states at intermediate time points. In radiation and G-CSF treatment scenarios, only endothelial cell data was used for training, generating perturbed transcriptomes for all three cell types. Finally, the biological significance of the predicted results was validated through differential expression and pseudo-temporal analysis.
Squidiff: A Conditional Diffusion Model Integrating DDIM and Semantic Encoding
To accurately predict the dynamic response of the transcriptome under various perturbations such as differentiation, development, gene editing, and drug treatment, the research team developed Squidiff, an intelligent computing framework based on the conditional diffusion model.As shown in the figure below, this model deeply integrates the Conditional Denoising Diffusion Implicit Model (DDIM) with semantic encoding technology to construct a three-stage collaborative architecture of "encoding-diffusion-decoding". It can not only efficiently generate transcriptome data that conforms to the biological background, but also flexibly regulate cell state through latent variables, and is widely applicable to various research scenarios such as cell differentiation, gene perturbation and drug treatment.
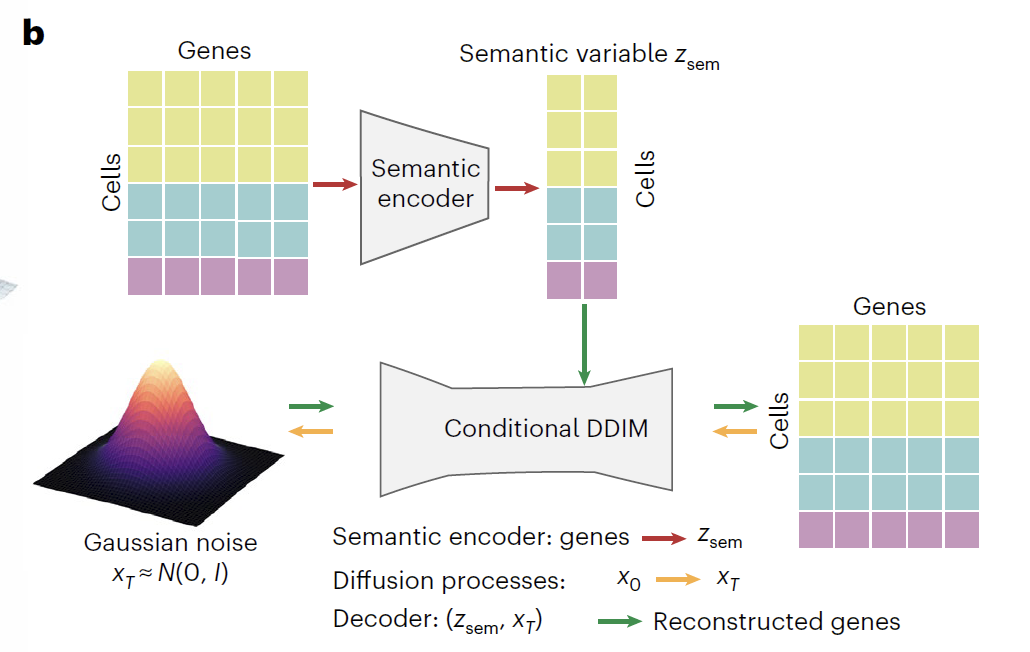
The core of Squidiff consists of a semantic encoder and a conditional DDIM diffusion module. The semantic encoder uses a multilayer perceptron (MLP) to map single-cell RNA sequencing data into a low-dimensional semantic space, generating semantic variables (Z_sem) containing cell type and perturbation information. For drug research scenarios, this encoder integrates recalibrated functional class fingerprints (r_FCFP), encoding the drug molecular structure as a 2,048-dimensional vector embedded in the semantic space. To predict unknown drug perturbations, the model also includes an adapter module that supports inputting drug SMILES strings and dosage information, achieving a deep fusion of biological and chemical information.
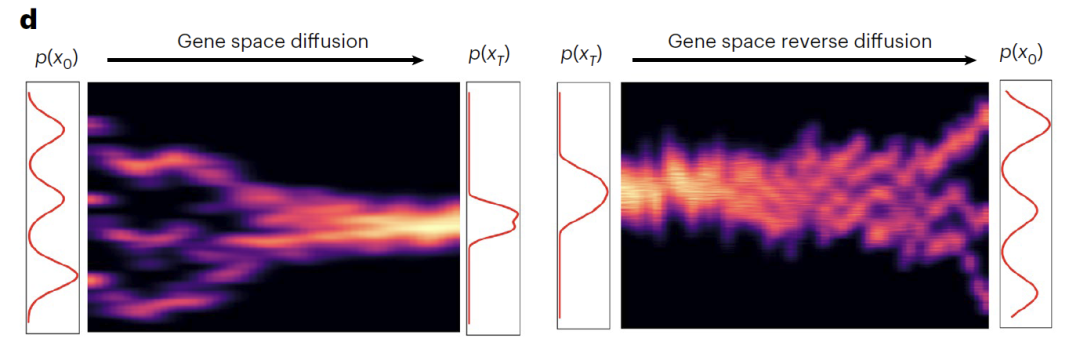
The conditional DDIM module follows a dual-process design of forward diffusion (Gene space diffusion) and reverse diffusion (Gene space reverse diffusion).During the forward diffusion process, the original gene expression data (x₀) is gradually transformed into approximately pure noise (x₀) through 1,000 iterations.In this process, the three typical cell types gradually approach a Gaussian distribution, while Z_sem effectively captures the biological variations in gene expression, clearly separating different experimental conditions in the latent space. During the backdiffusion process, a noise prediction network equipped with sinusoidal position embedding (ε) is used.Using time step (t) and Z_sem as dual conditions, the biologically significant transcriptome was reconstructed from x_T through iterative denoising, successfully restoring the original transcriptome profile.
The model training focuses on noise prediction loss as the core optimization objective, employs the Adam optimizer (learning rate 1×10⁻⁴) and relies on GPU acceleration.By coordinating the regulation of time steps and semantic variables, the model can simulate the continuous evolution of cell states, providing support for dynamic trajectory prediction.
Compared to traditional variational autoencoders, Squidiff has significant advantages:Without requiring the assumption of Gaussian distribution, it captures complex gene expression patterns through fine noise reduction, improving the F1 score by 27% in the prediction of rare cell types (<5%). It innovatively introduces the "gradient interpolation" strategy, which generates continuous differentiation paths by linearly combining semantic variables in the latent space, and successfully identifies transient cell states that are easily missed by traditional models (such as mesoderm precursors in iPSC differentiation).
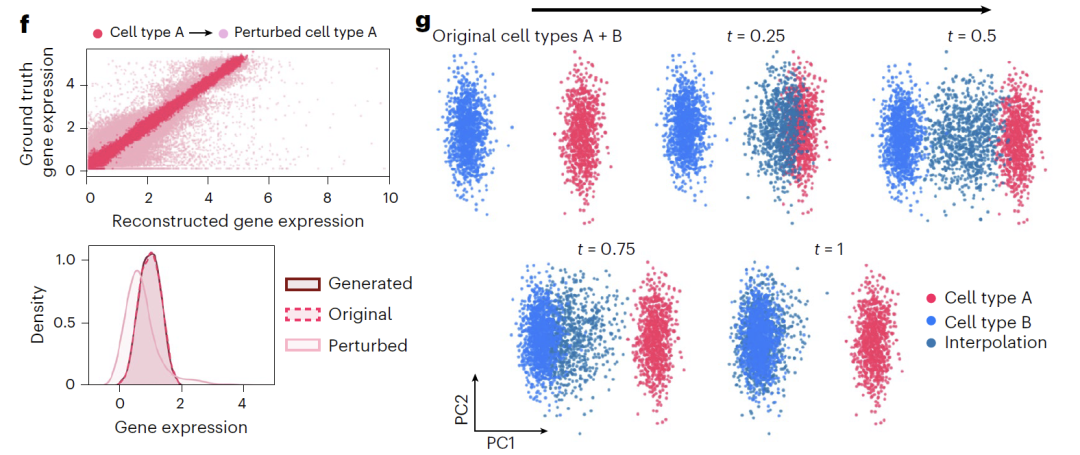
In addition, the model provides two methods for manipulating latent variables: "addition" combines the original representation with the perturbation direction (Δz_sem), as shown in Figure f below, to shift the gene expression distribution and reflect the perturbation effect; "interpolation" uses linear interpolation, as shown in Figure g below, to generate continuous states by obtaining intermediate points on the vector connection line, thus achieving a smooth transition of cell types.
Squidiff Multi-Scenario Demonstration: Precisely Capturing Transcriptomic Changes in Cell Differentiation, Perturbation, and Radiation Response
To systematically verify the transcriptome prediction capabilities of Squidiff, the research team conducted experimental verification in four key areas: cell differentiation, gene and drug perturbation, vascular organoid development, and radiation damage.
In cell differentiation prediction, as shown in the figure below, the team trained the model using only day 0 and day 3 data based on the iPSC-to-endoderm differentiation dataset. Differentiation direction was obtained by calculating semantic variable differences, and Squidiff successfully predicted the intermediate state between days 1 and 2. The model accurately captured the downregulation of the pluripotency marker MMOG, the upregulation of the endoderm factor GATA6, and identified the transient expression of the mesodermal marker DBX1. Compared with traditional methods, the transcriptome data generated by Squidiff can reconstruct a continuous path highly consistent with the actual developmental trajectory.
The model demonstrates outstanding performance in predicting gene and drug perturbations.For dual-gene knockout experiments in K562 cells, Squidiff can accurately predict non-additive effects without prior knowledge, and its robustness surpasses existing methods.In drug trials, the model was able to predict the synergistic effects of combination drugs using only single-drug data and accurately identify the specific effects of pabicept on tumor cells. Furthermore, by integrating a drug compound adapter, the model's predictive performance for the unknown drug sglt1 was comparable to that of specialized models, demonstrating excellent generalization ability.
In their research on vascular organoids (BVO), the team successfully predicted cell states at multiple intermediate time points using an iPSC-induced BVO model.The model not only reproduced the differentiation trajectories of the three main cell types—endothelial cells, fibroblasts, and mural cells—but also identified the intermediate state of mural cell differentiation into endothelial cells, which is difficult to capture using traditional methods. Gene expression analysis showed that the characteristic gene changes in the predicted data were highly consistent with known developmental patterns.
In radiation damage studies, the model accurately predicted the effects of radiation on various cell types using only endothelial cell training data. Analysis showed that early-developing cells were more sensitive to radiation, and the differentially expressed genes and related pathways predicted by the model were experimentally confirmed. In the prediction of G-CSF protective effects, the model revealed the drug's protective mechanisms against different cell types: activation of angiogenesis pathways in fibroblasts, inhibition of apoptosis pathways in endothelial cells, and enhanced genomic stability in wall cells. Experimental validation showed that cell death was significantly reduced after G-CSF treatment, demonstrating the reliability of the model's predictions.
These system experiments demonstrate that Squidiff can not only accurately predict changes in cell state under various biological scenarios, but also capture transient states and infer unknown perturbations, providing a powerful and reliable computational tool for predicting cell responses.
AI-driven new paradigm for single-cell research
In the interdisciplinary field of single-cell biology and artificial intelligence, the breakthrough in diffusion model technology represented by Squidiff is driving collaborative innovation between academia and industry.
At the academic research level, top university teams around the world continue to make breakthroughs in the depth and breadth of single-cell modeling.A research team at the University of Toronto in Canada has developed and released scGPT, the first fundamental large-scale language model for single-cell biology.The model is based on a generative pre-trained Transformer architecture and was trained on over 33 million cell data points covering 51 human organs/tissues and 441 independent studies. It comprehensively covers multiple cell types and physiological and pathological states, and fully presents a rich atlas of human cell heterogeneity.
Paper title:scGPT: Towards Building a Foundation Model for Single-Cell Multi-omics Using Generative AI
Paper address:
https://biorxiv.org/content/10.1101/2023.04.30.538439
at the same time,The Stanford University team focused on innovation in the spatial dimension, developing the three-dimensional spatiotemporal modeling framework Spateo.Based on scalable and precise algorithms, this framework can reconstruct complete three-dimensional embryo and organ models from continuous two-dimensional tissue slice data, and build a multi-level spatial digital system from single-cell molecular features to macroscopic embryo morphology.
Paper title:Spatiotemporal modeling of molecular holograms
Paper address:
https://www.cell.com/cell/fulltext/S0092-8674(24)01159-0
The business community transforms these academic findings into practical tools, demonstrating their significant value in drug development, disease treatment, and other scenarios.Cell2Sentence-Scale 27B (C2S-Scale 27B), developed by Google in collaboration with Yale University and other institutions, is one of the world's largest basic models for single-cell analysis.This model, built upon the Gemma open-source model family, boasts 27 billion parameters and is capable of deeply analyzing gene expression patterns in individual cells and accurately predicting cellular responses to drug interventions. Currently, the model has been integrated into Google Health's drug screening platform, supporting the design of personalized combination therapies for "cold tumors" and accelerating the development of immunotherapy regimens.Another important practice comes from the collaboration between the non-profit Arc Institute and companies such as 10x Genomics, whose STATE model focuses on simulating dynamic cellular responses.It integrates observational data from 170 million cells and interventional data from 100 million cells, enabling precise simulation of transcriptomic changes in cells under drug treatment, gene editing, or radiation exposure.
It is not difficult to see that from the academic community's in-depth exploration of basic single-cell models to the industry's large-scale implementation of the technology,Squidiff's diffusion modeling technology is driving single-cell research from "analyzing cell state" to "predicting cell fate".This leap not only accelerates the progress in areas such as drug development and cancer treatment, but will also provide core technological support for future medical directions such as precision medicine and regenerative medicine, continuously unleashing the enormous potential of AI-driven innovation in life sciences.
Reference articles:
1.https://mp.weixin.qq.com/s/yCR_GC0Ln80st2tHcv08-Q
2.https://mp.weixin.qq.com/s/GegQB65w4nZG6ZXvnyU9dw








