Command Palette
Search for a command to run...
Durch Die Halbierung Der Rechenkosten „kodiert“ ChemOntology, Ein Werkzeug Zur Entdeckung Chemischer Reaktionen, Die Menschliche Intuition in Sein System Und Beschleunigt so Die Suche Nach Reaktionswegen.
Chemische Reaktionsmechanismen offenbaren nicht nur die grundlegenden Gesetze der Stoffumwandlung, sondern liefern auch entscheidende Erkenntnisse für industrielle Anwendungen wie die Entwicklung effizienter Katalysatoren und umweltfreundlicher Syntheseverfahren. Die Analyse von Reaktionsmechanismen stützt sich maßgeblich auf eine zentrale computergestützte Methode – die Suche nach Reaktionswegen. Dabei werden lokale Minima und Reaktionszwischenprodukte auf der Potenzialenergiefläche (PES) lokalisiert, um den tatsächlichen Reaktionsweg zu ermitteln.
Lange Zeit haben sich Computerchemiker hauptsächlich auf Methoden der intrinsischen Reaktionskoordinaten (IRC) gestützt, um Reaktionsmechanismen durch die Generierung endlicher Konfigurationen zu untersuchen. Dieser traditionelle Ansatz weist jedoch erhebliche Einschränkungen auf. Er ist oft durch den vorgezeichneten Forschungsweg begrenzt und neigt dazu, unkonventionelle Reaktionswege zu übersehen, wodurch potenziell alternative Mechanismen unentdeckt bleiben.
Mit der Entwicklung automatisierter Methoden wie Artificial Force-Induced Response (AFIR) ist die unvoreingenommene Suche nach Reaktionspfaden möglich geworden. Diese Methoden betrachten Reaktionspfade als ein Netzwerk miteinander verbundener „Knoten“ und untersuchen systematisch Reaktionsmöglichkeiten durch iterative Generierung neuer Konfigurationen. Dadurch eröffnet sich ein neues Fenster zur Entdeckung unbekannter Reaktionsmechanismen.
Die automatisierte Pfadfindung ist jedoch keine perfekte Lösung. Die Energieberechnungen für zahlreiche Konfigurationen sind sehr aufwendig, und die Notwendigkeit, die Mechanismen der Konformationsänderungen zu untersuchen, erhöht den Rechenaufwand zusätzlich. Obwohl semi-empirische Methoden und maschinelle Lernverfahren die Kosten teilweise senken können, können gelegentliche Ungenauigkeiten bei der Energievorhersage die Zuverlässigkeit der Pfadfindung beeinträchtigen.
Die chemische Ontologie, als „Werkzeug zur Wissensstrukturierung“, bietet einen neuen Ansatz zur Überwindung der genannten Engpässe. Durch standardisierte Definitionen von Entitäten, Attributen, Beziehungen und Regeln organisiert sie fragmentiertes chemisches Wissen in maschinenlesbare und verarbeitbare strukturierte Informationen. Ontologie-Frameworks wie RXNO haben beispielsweise ihren Nutzen bei der Annotation von Reaktionswegen bereits unter Beweis gestellt.
Darauf aufbauend hat ein Forschungsteam der Universität Hokkaido in Japan ein neuartiges KI-System namens ChemOntology entwickelt.Als System zur Klassifizierung chemischen Wissens formalisiert es das chemische Denken des Menschen in einen maschinenverständlichen Rahmen und ermöglicht so die schnelle Erforschung und Analyse chemischer Reaktionen.Die erfolgreiche Anwendung dieses Rahmens bei der Untersuchung des klassischen Heck-Reaktionsmechanismus bestätigt nicht nur seine Effektivität bei der Beschleunigung der Pfadsuche, sondern unterstreicht auch das enorme Potenzial der Integration von „menschlichem chemischem Wissen“ mit „automatisierter Berechnung“.
Die zugehörigen Forschungsergebnisse mit dem Titel „ChemOntology: Eine wiederverwendbare, explizite, auf chemischer Ontologie basierende Methode zur Beschleunigung der Reaktionswegsuche“ wurden in ACS Catalysis veröffentlicht.
Forschungshighlights
* Es gelingt ihm, die Intuition menschlicher Chemiker erfolgreich in das System zu "programmieren", ohne auf einen Trainingsdatensatz angewiesen zu sein, was einen erheblichen Vorteil gegenüber herkömmlichen Methoden des maschinellen Lernens darstellt;
* Experimentelle Ergebnisse zeigen, dass ChemOntology in Kombination mit AFIR Ergebnisse erzielen kann, die mit einer vollständigen AFIR_TARGET-Suche vergleichbar sind, wenn nur etwa die Hälfte der Pfade untersucht wird, wodurch sich die gesamten Rechenkosten um fast die Hälfte reduzieren.
Papieradresse:
https://pubs.acs.org/doi/10.1021/acscatal.5c06298
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „ChemOntology“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
https://hyper.ai/papers
Datenmethodik des wissensbasierten Rahmens
Die von diesem Forschungsinstitut genutzten Datenressourcen sind nicht die riesigen Datensätze, die üblicherweise zum Trainieren von Modellen des maschinellen Lernens verwendet werden. Dies liegt genau an den inhärenten Eigenschaften der ChemOntologie als wissensbasiertem Rahmenwerk: Sie konzentriert sich auf chemische Regeln und Mechanismen anstatt auf Datenanpassung und vermeidet so die starke Abhängigkeit von großen Datenmengen und deren potenzielle methodische Einschränkungen.
Erste,Die Forscher nutzten die öffentliche chemische Datenbank PubChem, um standardisierte Informationen über alle Schlüsselkomponenten der Reaktion zu erhalten.Dies umfasst Molekülstruktur, Name und eindeutige Kennung. Diese Informationen können als „Ausweis“ für jede chemische Substanz betrachtet werden. Sie helfen nicht nur, die Rolle jeder Komponente im Reaktionssystem genau zu definieren, sondern ermöglichen auch die Verfolgung von Zielprodukten und die Eliminierung irrelevanter oder unnötiger Nebenprodukte durch eindeutige Verbindungsnummern. Dadurch wird die Suche nach nachfolgenden Reaktionswegen genauer und effizienter.
Zweitens, um die Zuverlässigkeit und Anwendbarkeit der Methode in realen und komplexen chemischen Szenarien zu testen,Die Forscher wählten die klassische Heck-Reaktion, die verschiedene Mechanismen und zahlreiche Reaktionsschritte aufweist, als Testfall.Das System wurde mit vollständigen Eingangsdaten versorgt, darunter dreidimensionale Strukturdateien von Reaktanten, Katalysatoren, Liganden und Basen sowie Referenzenergiedaten für bekannte Zwischenprodukte und Endprodukte. Dieser repräsentative Fall untersucht die Leistungsfähigkeit der Methode in komplexen Reaktionsnetzwerken umfassend und bestätigt nicht nur ihre Fähigkeit, wichtige Zwischenprodukte zu identifizieren und zwischen Haupt- und Nebenreaktionswegen zu unterscheiden, sondern demonstriert auch anschaulich ihre Vorteile bei der Reduzierung des Rechenaufwands.
Insgesamt gewährleistet diese Studie die Genauigkeit der Informationen durch maßgebliche Datenbanken, überprüft die Effektivität der Methoden durch die Nutzung typischer komplexer Reaktionen und fördert Zusammenarbeit und Iteration durch vollständige Open-Source-Nutzung, wodurch eine breite Anwendbarkeit auf diverse metallorganische Reaktionssysteme ohne die Notwendigkeit umfangreicher Trainingsdaten gewährleistet wird.
ChemOntologie: Ein neues Rahmenwerk zur Suche nach Reaktionswegen in metallorganischen Reaktionen
ChemOntology ist ein wissensbasiertes Rechenframework, dessen Kernidee nicht auf dem Training von Modellen mit umfangreichen Daten beruht.Stattdessen integriert es systematisch chemische Reaktionsregeln, strukturelle Einschränkungen und quantenchemische Suchprozesse für Reaktionswege.Dies ermöglicht die effiziente Untersuchung von Reaktionswegen innerhalb eines definierten chemischen Kontextes. Die Methode nutzt AFIR (Artificial Force Induced Reaction) als Rechenmodell, wobei chemisches Wissen explizit kodiert wird, um die Suchrichtung zu steuern, und ein Echtzeit-Screening der generierten Strukturen durchgeführt wird, um sinnlose oder unplausible Reaktionsverläufe zu vermeiden.
Wie in der folgenden Abbildung dargestellt, besteht der ChemOntology-Workflow aus der Verarbeitung der Benutzereingaben, der Modellierung chemischer Informationen in der Setup-Datei, der Generierung von Reaktionswegen mithilfe von ERPOs, strukturellen Rationalitätsbeschränkungen, der Ausführung und Steuerung von AFIR sowie der Pfadanalyse.
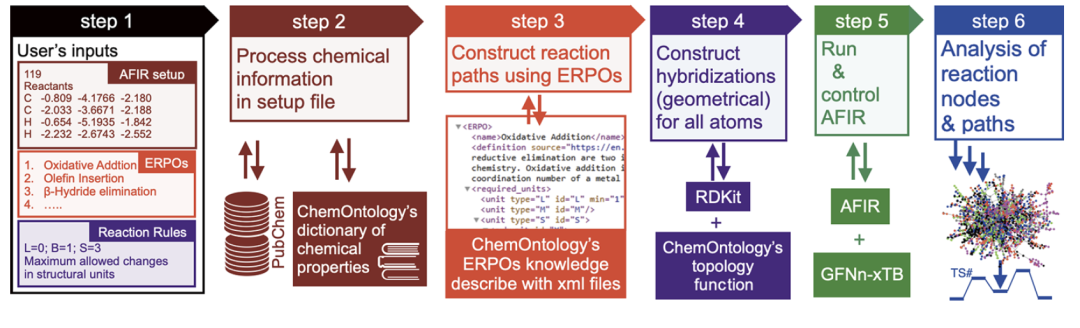
Das Reaktionssystem wird zunächst als eine Sammlung von Struktureinheiten wie Metallen, Liganden, Substraten und optionalen Basen analysiert, wobei jeder Einheitenart eine spezifische chemische Rolle und Eigenschaft zugewiesen wird.Der Reaktionsprozess wird als schrittweise Transformation der Hybridisierungszustände von Struktureinheiten und ihren internen Atomen beschrieben, wodurch Strukturänderungen auf drei Ebenen nachvollzogen werden: „Reaktionsknoten – Struktureinheit – Atom“. Diese hierarchische Darstellung ermöglicht es dem Modell, die chemische Plausibilität von Reaktionswegen allein anhand geometrischer und topologischer Informationen zu bestimmen, ohne auf Details der elektronischen Struktur angewiesen zu sein.
Die Generierung von Reaktionswegen basiert auf ERPO (Elementarer Reaktionswegoperator).Das heißt, eine modulare Beschreibung gängiger metallorganischer Elementarreaktionsprozesse.Beispiele für Reaktionen sind die Bildung von Koordinationsverbindungen, oxidative Addition, Olefininsertion und β-Wasserstoffeliminierung. ERPO dient nicht nur der Konstruktion von Reaktionssequenzen, sondern auch der Regelverifizierung während des Suchprozesses und stellt sicher, dass jede Strukturtransformation der erwarteten chemischen Semantik entspricht. Durch die Zerlegung komplexer Reaktionen in kombinatorische Elementarprozesse kann ChemOntologie die kombinatorische Komplexität des Suchraums deutlich reduzieren und gleichzeitig die Reaktionsvielfalt erhalten.
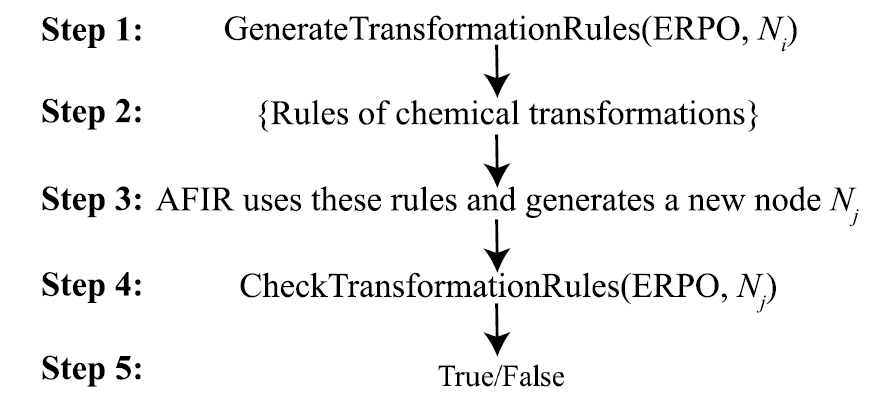
Um die Reaktionsentwicklung weiter einzuschränken,ChemOntology führt einen Filtermechanismus ein, der auf Veränderungen der atomaren Hybridisierung basiert.Mithilfe weniger Parameter können Anwender die maximal zulässigen Strukturanpassungen für verschiedene Struktureinheiten während des Reaktionsprozesses begrenzen. Geometrische Strukturen, die die Beschränkungen überschreiten, werden automatisch erkannt und aus der Suche entfernt. Dieser Mechanismus unterdrückt effektiv das Problem der Strukturexplosion und verbessert die Recheneffizienz erheblich, ohne dass spezifische Reaktionsergebnisse vorab festgelegt werden müssen.
In der praktischen Berechnung wird ChemOntology als Wissenskontrollschicht über dem AFIR-Suchprozess eingebettet und mit der semi-empirischen Tight-Binding-Methode GFN2-xTB kombiniert, um die geometrische Entwicklung des Reaktionswegs zu beschreiben. Im Gegensatz zu Modellen des maschinellen LernensChemOntology benötigt kein Training mit einem Datensatz; seine "Wissensbasis" besteht hauptsächlich aus Regeln zur Erkennung funktioneller Gruppen, Klassifizierungsschemata für Struktureinheiten und ERPO-Dateien.Alle diese Parameter lassen sich vom Benutzer flexibel an das jeweilige Forschungsobjekt anpassen. Dadurch ähnelt ChemOntology eher einer computergestützten chemischen Methodik, die dazu dient, menschliche chemische Intuition systematisch in den automatisierten Prozess der Reaktionserkundung einzubringen.
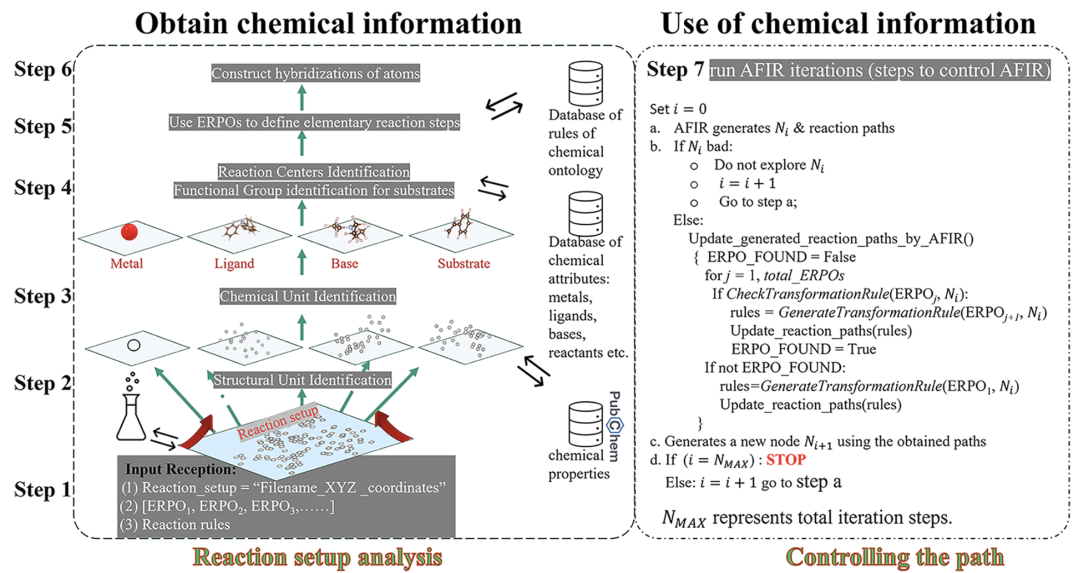
Insgesamt bietet ChemOntology eine Plattform zur Suche nach Reaktionswegen unter expliziten chemischen Randbedingungen: Sie schränkt nicht das Auftreten neuer Reaktivitäten ein, sondern lenkt Berechnungen so, dass sie innerhalb eines „vernünftigen chemischen Raums“ durch strukturierte Regeln forschen und so ein Gleichgewicht zwischen der Analyse von Reaktionsmechanismen und potenziellen neuen chemischen Entdeckungen erreichen.
Experimentelle Ergebnisse: Rechenaufwand halbiert, Pfadübersichtlichkeit verdoppelt.
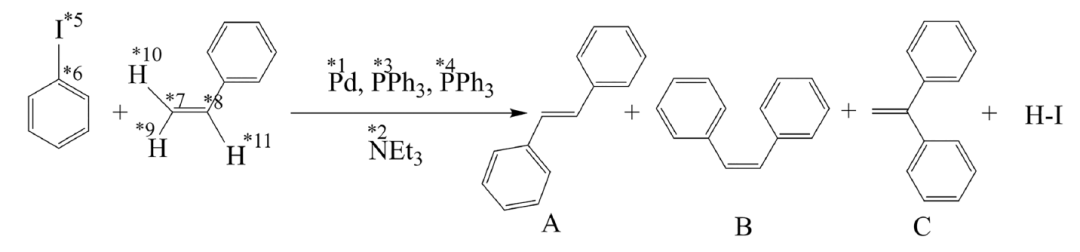
Um die Effektivität und Effizienz des ChemOntology-Frameworks bei der Suche nach Reaktionswegen zu überprüfen,Das Forschungsteam wählte die klassische Heck-Reaktion, die einen komplexen Mechanismus aufweist und repräsentativ ist, als Testsystem.Wie in der Abbildung unten dargestellt, werden bei dieser Reaktion Iodbenzol und Styrol als Substrate verwendet. Unter Palladiumkatalyse, mit Triphenylphosphin als Ligand und Triethylamin als Base entsteht hauptsächlich trans-Stilben, begleitet von geringen Mengen des cis-Isomers und Spuren von Nebenprodukten. Der Reaktionsmechanismus umfasst mehrere Schlüsselschritte, darunter oxidative Addition, Olefininsertion, migratorische Insertion, β-Wasserstoffeliminierung und Baseneliminierung. Die zahlreichen Reaktionszentren stellen eine typische Herausforderung für automatisierte Methoden zur Reaktionsfindung dar.
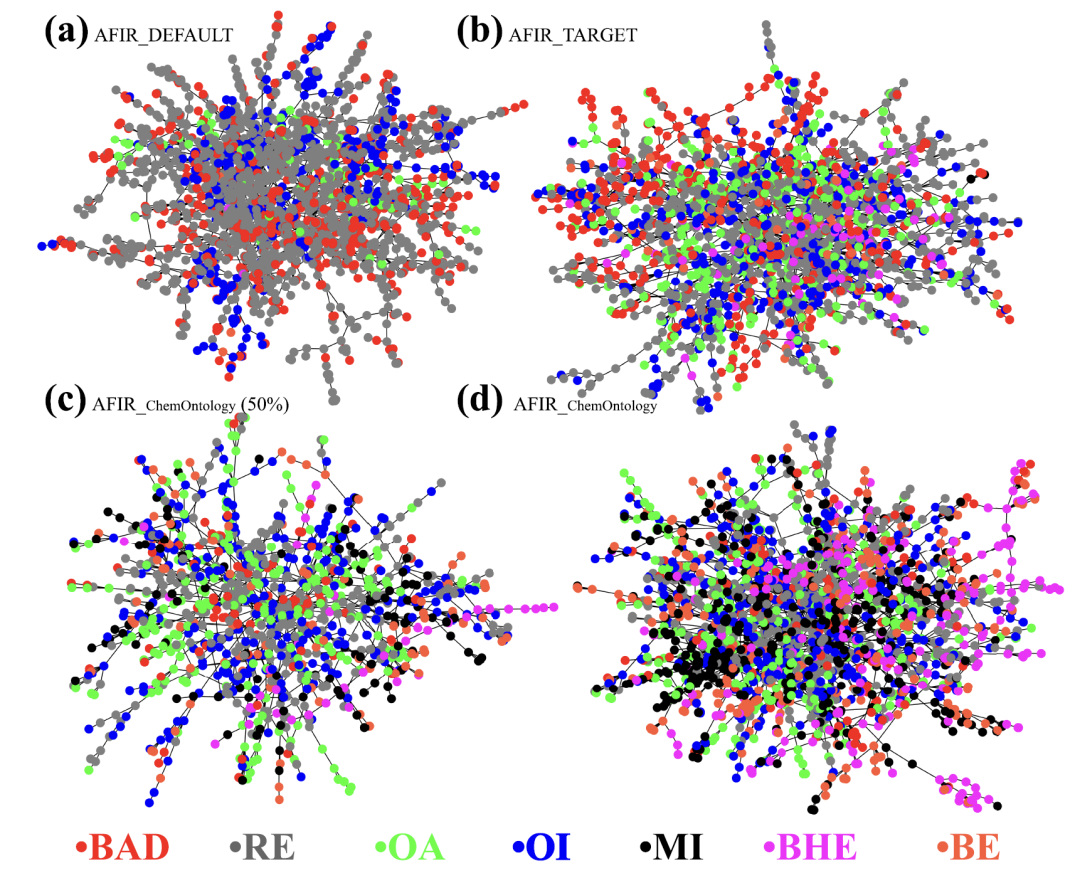
Die Studie verglich drei parallele Pfadsuchstrategien: die ungelenkte Strategie AFIR_DEFAULT, die teilweise eingeschränkte Strategie AFIR_TARGET und AFIR_ChemOntology, die eine chemische Ontologie einbezieht. Diese drei Strategien unterscheiden sich grundlegend in ihrem Grad an „Intelligenz“: Die erstgenannte durchsucht den Konfigurationsraum nahezu wahllos, während die letztgenannte den Suchraum durch künstliche Einschränkungen begrenzt.AFIR_ChemOntology hingegen identifiziert mithilfe seines Frameworks automatisch die chemischen Rollen von Reaktanten und wichtigen Reaktionszentren und steuert die Suche dynamisch durch die Nutzung elementarer Reaktionsprozesse.
Unter gleichen Rechenbedingungen unterscheiden sich die von den drei Methoden generierten Reaktionsnetzwerke, wie in der Abbildung unten dargestellt, deutlich. AFIR_DEFAULT erzeugt eine große Anzahl chemisch bedeutungsloser, ungültiger Knoten, wodurch die effektiven Pfade stark überlastet werden; AFIR_TARGET zeigt zwar eine gewisse Verbesserung, weist aber immer noch viele redundante Strukturen auf; im Gegensatz dazuDie Suchergebnisse für AFIR_ChemOntology sind hochgradig zielgerichtet und ermöglichen die frühzeitige und eindeutige Identifizierung wichtiger Reaktionswege.Die Berechnungen konzentrierten sich auf chemisch plausible Reaktionswege. Weitere Zwischenstatistiken zeigten, dass ChemOntology den Anteil fehlerhafter Knotenpunkte deutlich reduzierte und die identifizierten Schlüsselzwischenprodukte weitgehend mit dem klassischen Mechanismus der Heck-Reaktion übereinstimmten.
Wie aus der untenstehenden Abbildung hervorgeht, zeigt die Energieanalyse, dass alle drei Methoden einen gemeinsamen Schritt in den frühen Stadien der Reaktion erfassen.Allerdings kann nur AFIR_ChemOntology die spezifischen Wege, die zum Hauptprodukt bzw. zum Nebenprodukt führen, vollständig unterscheiden und verfolgen.Darüber hinaus wurden im effizienten Reaktionsweg charakteristische Wechselwirkungen beobachtet, die mit der β-Wasserstoffeliminierung verbunden sind, während im Reaktionsweg, der zu Spurenprodukten führt, diese Wechselwirkungen eine geringere strukturelle Stabilität aufwiesen, was ihre niedrigere Entstehungswahrscheinlichkeit erklären könnte.
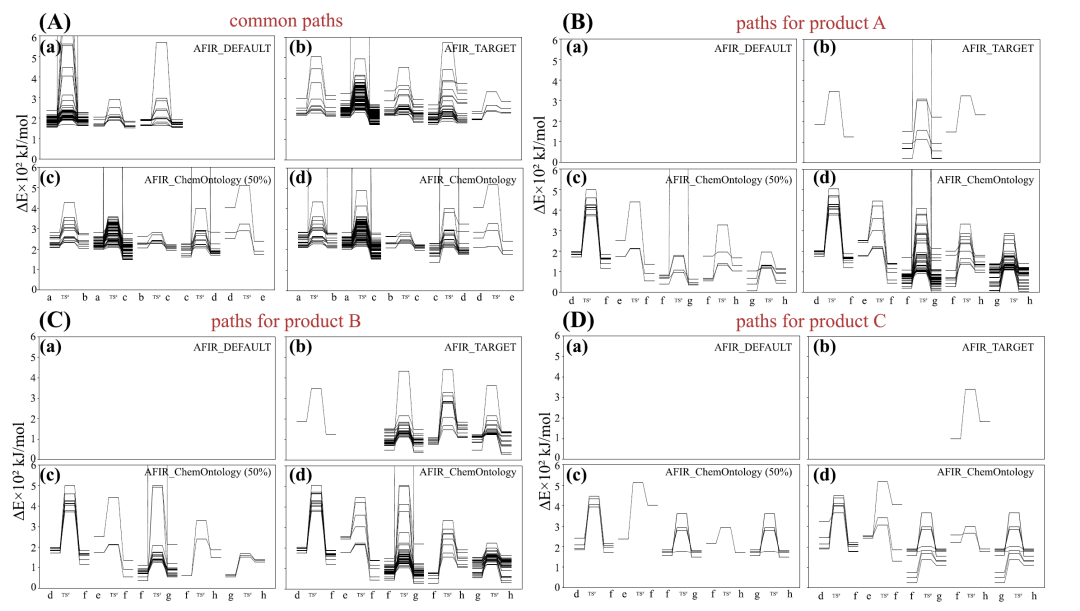
Im Hinblick auf die Recheneffizienz,AFIR_ChemOntology erzielt eine vergleichbare Effizienz wie eine vollständige Suche von AFIR_TARGET, während nur etwa die Hälfte der Pfade durchsucht wird, wodurch die gesamten Rechenkosten um fast die Hälfte reduziert werden.Dieser Vorteil beruht primär auf der Nutzung chemischer Kenntnisse zur Steuerung der Suchrichtung und der Echtzeitfilterung ungültiger Strukturen. Insgesamt zeigen die experimentellen Ergebnisse, dass die Integration chemischer Ontologie in die automatisierte Pfadsuche die Effizienz mechanistischer Analysen deutlich verbessern und gleichzeitig die chemische Rationalität gewährleisten kann. Dies bietet einen effizienteren und zuverlässigeren Ansatz zur Untersuchung komplexer Reaktionssysteme.
Vom Labor zur Fabrik: Neugestaltung des Weges der Reaktionsforschung durch chemische Ontologie
Die Integration chemischer Ontologie und automatisierter Reaktionswegsuche schlägt eine entscheidende Brücke zwischen theoretischer Chemie und industriellen Anwendungen. Dieser Trend hat nicht nur eine Reihe bahnbrechender Forschungen in der Wissenschaft angestoßen, sondern auch bedeutende innovative Praktiken in der Industrie hervorgebracht und die Forschung zu Reaktionsmechanismen von der traditionellen „nachträglichen Analyse“ hin zu einer proaktiven „aktiven Steuerung“ transformiert.
In der akademischen Forschung liegt der Fokus auf algorithmischer Innovation und der Verfeinerung von Mechanismen, wodurch die Grenzen des Wissens auf diesem Gebiet kontinuierlich erweitert werden. So entwickelte beispielsweise ein Team der Universität Island den Algorithmus „Optimal Transport Gaussian Process“ (OT-GP).Kernstück ist die Anwendung einer intelligenten Datenfilterstrategie, die auch mit einer begrenzten Menge an Trainingsdaten effizient arbeitet.Dieser Algorithmus reduziert die durchschnittliche Suchzeit für molekulare Reaktionswege signifikant von 28,3 Minuten auf 12,6 Minuten und verbessert die Erfolgsquote deutlich. Er bietet somit ein neues Werkzeug zur schnellen Erforschung von Mechanismen komplexer Systeme.
Titel des Papers: Adaptives Pruning zur Erhöhung der Robustheit und Reduzierung des Rechenaufwands bei Gaußprozessbeschleunigten Sattelpunktsuchen
Link zum Artikel:https://doi.org/10.48550/arXiv.2510.06030
gleichzeitig,Ein Forschungsteam der ETH Zürich in der Schweiz kombinierte Ab-initio-Molekulardynamik mit verbesserten Sampling-Methoden.Wir haben systematisch die wichtigsten Schritte des Wasserstofftransfers und der Umlagerung in der katalytischen Reaktion von Molekularsieben und Übergangsmetallen untersucht, die Mechanismencharakteristika der dynamischen Veränderungen der Reaktionskanäle in Abhängigkeit von der Reaktionsumgebung aufgezeigt und ein allgemeines mikroskopisches Bild vorgeschlagen, das als Leitfaden für die rationale Entwicklung von Katalysatoren dienen kann.
Titel der Arbeit: Ab-initio-Molekulardynamik mit verbessertem Sampling in der heterogenen Katalyse
Link zum Artikel:https://pubs.rsc.org/en/content/articlelanding/2022/cy/d1cy01329g
Die Industriepraxis hingegen konzentriert sich stärker auf die Umsetzung dieser Theorien in praktische Produktivität. Nehmen wir beispielsweise Schrödinger, ein führendes Unternehmen im Bereich der Computerchemie in den Vereinigten Staaten.Der automatisierte Reaktionsworkflow AutoRW integriert die strukturierte Denkweise der chemischen Ontologie auf tiefgreifende Weise.Es erreicht eine vollständige Prozessautomatisierung von der Reaktionsaufzählung und Pfadabbildung bis hin zur Ergebnisorganisation und -ausgabe.
Die Zusammenarbeit zwischen dem deutschen Chemiekonzern BASF und IBM zeigt unterdessen ebenfalls einen ähnlichen Weg der technologischen Integration auf.Beide Parteien werden chemische Ontologie mit quantenchemischen Berechnungen und künstlicher Intelligenz kombinieren, um gemeinsam die Forschung und Entwicklung von Hochleistungskatalysatoren voranzutreiben.Durch die Anwendung des Modells „wissensgeleitetes Rechnen mit KI“ konnte nicht nur der F&E-Zyklus deutlich verkürzt und die Kosten für experimentelles Ausprobieren reduziert werden, sondern es wurde auch eine solide Grundlage für die Anwendung von Polyurethanmaterialien in der Automobilindustrie, im Bauwesen und anderen Bereichen geschaffen.
Diese Praktiken führender globaler Unternehmen bestätigen nicht nur den universellen Wert der Kombination von chemischer Ontologie mit automatisierter Datenverarbeitung, sondern haben durch regionen- und domänenübergreifende technologische Zusammenarbeit auch einen positiven Kreislauf von akademischen Durchbrüchen über den Technologietransfer bis hin zu industriellen Anwendungen und Nachfragerückmeldungen geschaffen, der die globale Chemieindustrie kontinuierlich in Richtung einer grüneren, effizienteren und intelligenteren Zukunft treibt.
Referenzlinks:
1.https://wp-stg.schrodinger.com/wp-content/uploads/2023/10/A4-22_111-Reaction-Workflow-Application-Note_R3-1-1.pdf
2.https://blog.csdn.net/cainiao080605/article/details/147259567
3.https://phys.org/news/2025-12-ai-mimics-human-intuition-explore.html








