Command Palette
Search for a command to run...
Von 100.000 Möglichkeiten Bis Zur Erfolgreichen Synthese: Die Innovative Integration Globaler Aufmerksamkeitsmechanismen Und Des KI-Modells CGformer Erleichtern Die Entwicklung Hochentropischer Materialien.

Künstliche Intelligenz verändert das Paradigma der materialwissenschaftlichen Forschung und Entwicklung grundlegend und erweist sich als bahnbrechender Beitrag zur Beschleunigung der Entdeckung neuer Materialien und zur Optimierung ihrer Leistungsfähigkeit. Durch die tiefe Integration von Hochdurchsatz-Computing und maschinellem Lernen werden die Schwachstellen traditioneller „Versuch-und-Irrtum“-Methoden, wie lange Versuchszyklen und hoher Ressourcenverbrauch, effektiv behoben.Die Materialforschung ist in die effiziente iterative Phase der „computergestützten experimentellen Verifizierung“ eingetreten.Mit der Innovation menschlicher Technologie und Lebensweise werden jedoch die Leistungsanforderungen an neue Materialien in Bereichen wie neue Energien und Luft- und Raumfahrt immer strenger, und die Grenzen traditioneller Methoden des maschinellen Lernens werden allmählich offensichtlich, insbesondere im Bereich der Forschung und Entwicklung von Materialien mit hoher Entropie.
Hochentropiematerialien sind eine neue Werkstoffklasse, die aus einer Mischung mehrerer Hauptelemente besteht. Durch die synergistische Wirkung dieser Elemente erhöhen Hochentropiematerialien die Konfigurationsentropie (d. h. die Unordnung) ihrer Atomanordnung deutlich, was zu überlegenen mechanischen, temperaturbeständigen und korrosionsbeständigen Eigenschaften im Vergleich zu herkömmlichen Materialien führt. Diese Materialien bieten ein erhebliches Anwendungspotenzial in der Energiespeicherung, der Luft- und Raumfahrt sowie in Geräten für extreme Umgebungen.
Frühere Ansätze, wie Crystal Graph Convolutional Neural Networks (CGCNN) und Atomistic Line Graph Neural Networks (ALIGNN), weisen alle architektonische Mängel auf.Aufgrund der Beschränkung auf den lokalen Informationsinteraktionsmechanismus ist es schwierig, den synergistischen Effekt der Atome über große Entfernungen zu modellieren. Zudem können die für komplexe Kristallstrukturen einzigartigen globalen Informationen nicht vollständig erfasst werden, was zu einer eingeschränkten Vorhersagegenauigkeit führt.Gleichzeitig stellen die inhärenten Eigenschaften von Materialien mit hoher Entropie auch Herausforderungen in Bezug auf ihre Forschung und Entwicklung dar, die weit über die herkömmlicher Materialien hinausgehen.Komplexe Mikrostrukturen, wenige experimentelle Daten hoher Qualität und dynamisch ungeordnetes Atomverhalten stellen zusammen die Haupthindernisse bei der Entwicklung von Materialien mit hoher Entropie dar.
Als Reaktion auf Werkzeugdefekte und Nachfrage-Upgrades,Das Team von Professor Li Jinjin und Professor Huang Fuqiang vom Artificial Intelligence and Microstructure Laboratory (AIMS-Lab) der Shanghai Jiao Tong University hat ein neues KI-Materialdesignmodell namens CGformer entwickelt, das die Beschränkungen traditioneller Modelle erfolgreich durchbricht.Dieses Modell kombiniert auf innovative Weise den globalen Aufmerksamkeitsmechanismus von Graphormer mit CGCNN und integriert Zentralitätskodierung und räumliche Kodierung, sodass es nicht nur die Materialstruktur intuitiv durch Kristalldiagramme beschreiben kann, sondern mithilfe des Mechanismus der „globalen Aufmerksamkeit“ auch die Wechselwirkungen zwischen weit entfernten Atomen erfassen kann. Dadurch erhält es globale Informationsverarbeitungsfähigkeiten, die herkömmliche Modelle, die sich „nur auf benachbarte Atome konzentrieren“, nicht haben.
Diese Methode liefert umfassendere Strukturinformationen, ermöglicht eine genauere Vorhersage der Ionenmigration innerhalb von Strukturen und stellt ein zuverlässiges Werkzeug für die Forschung und Entwicklung neuer Materialien, insbesondere hochentropischer und komplexer kristalliner Materialien, dar. Die Forschungsergebnisse wurden in der renommierten Fachzeitschrift Matter unter dem Titel „CGformer: Transformer-enhanced crystal graph network with global attention for material property prediction“ veröffentlicht.
Forschungshighlights:
* Die Forschung und Entwicklung von CGformer, einem KI-Materialdesignmodell, das auf einem globalen Aufmerksamkeitsmechanismus basiert, bietet ein zuverlässiges und leistungsstarkes Werkzeug für die Materialforschung und -entwicklungswissenschaft und trägt dazu bei, den Entdeckungsprozess komplexer Kristallstrukturen zu beschleunigen.
* Im Vergleich zu CGCNN weist CGformer bei der Untersuchung von Natriumionen-Festelektrolyten mit hoher Entropie (HE-NSEs) eine Reduzierung des mittleren absoluten Fehlers um 25% auf, was seine Praktikabilität und Weiterentwicklung eindrucksvoll demonstriert.
*Aus 148.995 möglichen Strukturen mit hoher Entropie wurden 18 ausgewählt und sechs feste Natriumionenelektrolyte mit hoher Entropie (HE-NSEs) erfolgreich synthetisiert und verifiziert, mit einer Natriumionenleitfähigkeit von bis zu 0,256 mS/cm bei Raumtemperatur, wodurch ihr praktischer Anwendungswert nachgewiesen wurde.

Papieradresse:
https://www.cell.com/matter/abstract/S2590-2385(25)00423-0
Folgen Sie dem offiziellen Konto und antworten Sie mit „CGformer“, um das vollständige PDF zu erhalten
Weitere Artikel zu den Grenzen der KI:
Multikategorische Datensätze verbessern die Fähigkeiten des CGformer-Modells
Ziel dieser Studie ist es, die Herausforderungen, die sich aus Datenknappheit und struktureller Komplexität in Systemen mit hoher Entropie ergeben, durch fallbasierte Lösungen anzugehen.Der Forschungsfall konzentriert sich auf Elektrofahrzeuge mit neuer Energie und Anwendungen zur Netzenergiespeicherung, insbesondere auf die Leistungsvorhersage und das Screening von Natriumionen-Festelektrolyten mit hoher Entropie. Verschiedene Datensätze wurden erstellt und zur Unterstützung des Trainings, der Feinabstimmung und der experimentellen Verifizierung des CGformer-Modells verwendet.
Basisdatensatz der Natriumionendiffusionsenergiebarriere (Eb):Dies ist der größte bekannte Datensatz zu Natriumionendiffusionsbarrieren in Hochentropiestrukturen, der von Forschern für diese Studie erstellt wurde. Er basiert auf den Methoden der kristallographischen Analyse durch Voronoi-Zerlegung (CAVD) und der Bindungsvalenzstellenenergie (BVSE). Dieser Datensatz wird hauptsächlich für das Vortraining von CGformer verwendet. Dadurch kann das Modell Grapheninformationen zu natriumhaltigen Strukturen erlernen, die dann in den berechneten Hochentropiedatensatz übertragen werden und die Grundlage für die anschließende Eb-Vorhersage von Hochentropiematerialien bilden.
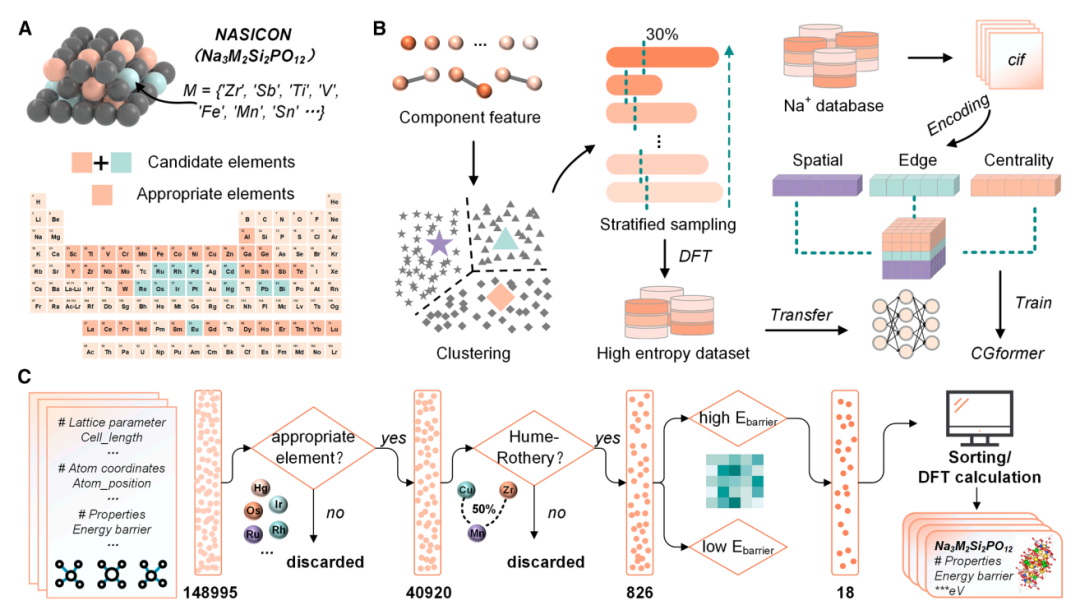
HE-NSEs-Berechnungsdatensatz:Basierend auf Na₃Zr₂Si₂PO₁₂ (wie oben gezeigt) wurden 45 potenzielle hochentropische Dotierungselemente am Zr-Standort berücksichtigt, was zu einem anfänglichen chemischen Raum mit 148.995 möglichen hochentropischen Strukturen führte. Anschließend wurde der chemische Raum durch mehrere Screening-Runden, einschließlich des Ausschlusses ungeeigneter Elemente (radioaktiv, hochgiftig und teuer) und Einschränkungen hinsichtlich Atomradiusunterschieden und Ladungsgleichgewicht, weiter auf 826 relativ stabile Strukturen eingegrenzt. Anschließend wurden diese Strukturen durch unüberwachtes hierarchisches Clustering in 20 Gruppen eingeteilt. Aus jeder Gruppe wurden 30%-Strukturen (insgesamt 238) hierarchisch ausgewählt und ihre Eb-Werte mithilfe der Dichtefunktionaltheorie (DFT) berechnet. Dies bildete letztendlich einen dedizierten Datensatz für die Feinabstimmung von CGformer, wobei das Modell speziell an die Aufgabe angepasst wurde, den Eb von Natriumionen in NASICON-Strukturen mit hoher Entropie vorherzusagen und so die Genauigkeit des Modells im Zielszenario zu verbessern.
Datensatz zur Bewertung der thermischen Stabilität:Die Forscher extrahierten alle natriumhaltigen Strukturen mit Energien oberhalb des konvexen Hüllenwerts (Ehull) aus der Materials Project-Datenbank und fassten sie in einem speziellen Trainingsdatensatz zusammen. Dieser Datensatz dient primär zum Training eines ergänzenden Modells zur Bewertung der thermodynamischen Stabilität von HE-NSEs. In Kombination mit dem von CGformer vorhergesagten Eb kann dieses Modell verwendet werden, um Kandidatenmaterialien auf hohe Leistung und Stabilität zu prüfen.
Innovative Fusionsarchitektur ermöglicht CGformers „globale Wahrnehmung“
CGformer hat grundlegende Innovationen entwickelt, um die Mängel herkömmlicher Methoden zu beheben, indem zwei fortschrittliche Technologien integriert wurden, um komplementäre Vorteile zu erzielen.Sein Kern besteht darin, die grafische Darstellungsfähigkeit der Kristallstruktur beizubehalten und die Beschränkung der Konzentration nur auf lokale atomare Wechselwirkungen durch den globalen Aufmerksamkeitsmechanismus aufzuheben.Insbesondere kombiniert es den globalen Aufmerksamkeitsmechanismus von Graphormer mit der Kristallgraph-Darstellungsmethode von CGCNN und fügt gleichzeitig wichtige Codierungsmodule hinzu, um eine neue Informationsverarbeitungspipeline aufzubauen.
Abbildung a unten zeigt den Kristalldiagramm-Kodierungsprozess.Der Prozess besteht darin, die reale dreidimensionale Kristallstruktur in ein Kristalldiagramm umzuwandeln, das vom Modell verarbeitet werden kann.Atome in einer Kristallstruktur werden als Knoten und die chemischen Bindungen zwischen den Atomen als Kanten dargestellt. Durch diesen Konvertierungsprozess konnten die Forscher Knoten- und Kantenmerkmale wie verschiedene Elementeigenschaften, Ladung, kovalenten Radius, interatomare Abstände, Bindungstypen und Informationen zur Kristallsymmetrie extrahieren. Diese Merkmale wurden dann kombiniert, um die von CGformer benötigten anfänglichen Eingabedaten zu erhalten. Dadurch wurde sichergestellt, dass die chemischen und strukturellen Informationen des Kristalls vollständig erhalten blieben.
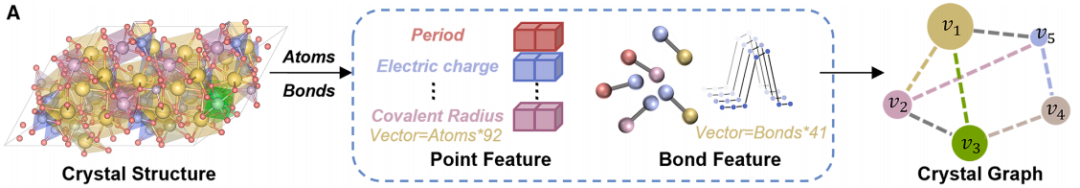
Abbildung b unten zeigt die Netzwerkarchitektur von CGformerDurch die Zusammenarbeit mehrerer Module werden eine globale Informationsintegration und genaue Vorhersagen erreicht.Zunächst durchläuft der eingegebene Kristallgraph eine Reihe von Graph-Faltungsoperationen, um eine vereinfachte Graphstruktur zu erzeugen. Dadurch wird der Rechenaufwand nachfolgender Netzwerkschichten reduziert und der Trainingsprozess von CGformer beschleunigt. Darauf aufbauend berechnen die Forscher den Center-Code und aktualisieren die Knotenmerkmale des Kristallgraphen. Der Center-Code enthält den Eingangs- und Ausgangsgrad jedes Knotens, die dann in die ursprünglichen Knotenmerkmale integriert werden. Jeder Knoten durchläuft anschließend ein Multi-Head-Attention-Modul (Multi-Head-Attention-Modul), das variable Merkmale und räumliche Kodierung kombiniert, um die Positionsbeziehung zwischen den Knoten darzustellen. Der Center-Code wandelt die durchschnittlichen Merkmale benachbarter Knoten in eine Summenform um, während die räumliche Kodierung es dem Self-Attention-Mechanismus ermöglicht, benachbarte Knoten zu unterscheiden, eine effektive Nachrichtenaggregation zu fördern und die Informationsverbindungen zwischen verschiedenen Atomen zu verbessern. Schließlich durchläuft der Ausgabevektor die Prozesse „Pooling (Integration globaler Merkmale)“ und „Aktivierung (Funktionsoperation)“, um die endgültige Vorhersage der Materialeigenschaften abzuschließen.
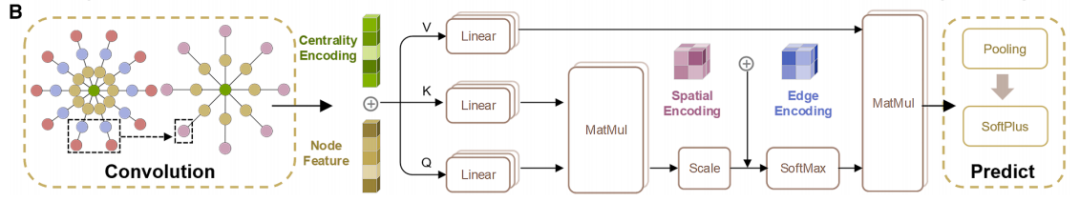
Das Multi-Head-Attention-Modul ermöglicht es jedem Knoten, nicht nur auf benachbarte Knoten, sondern auch auf alle anderen Knoten im Kristalldiagramm zu achten. Dadurch werden atomare Wechselwirkungen über große Entfernungen erfasst. Durch die zusätzliche Zentrums- und Raumkodierung kann das Modell nicht nur die chemischen Eigenschaften von Atomen identifizieren, sondern auch deren Positionsbedeutung und räumliche Beziehungen innerhalb der Struktur erkennen. Dies verbessert die Genauigkeit des Modells bei der Charakterisierung komplexer Kristalle.
insgesamt,Im Vergleich zu herkömmlichen Kristallnetzwerken hat CGformer einen qualitativen Sprung gemacht und drei wesentliche Vorteile realisiert: globale Vision, Informationsverbesserung und Effizienzbilanz, wodurch ein glaubwürdiges und zuverlässiges Tool für die Entdeckung und Leistungsoptimierung komplexer Materialien mit hoher Entropie bereitgestellt wird.
CGformer zeigt starke Leistung und unterstreicht seinen praktischen Orientierungswert
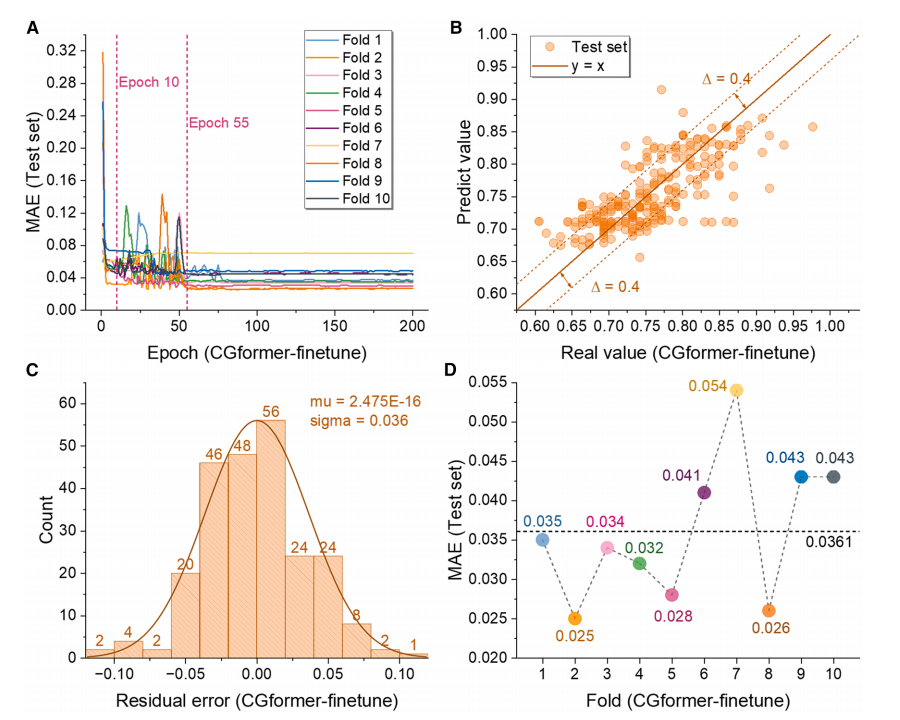
Um die Leistung und Weiterentwicklung des CGformer-Modells genau zu bewerten, verglichen die Forscher es mit traditionellen Modellen wie CGCNN, ALIGNN und SchNet. Das Experiment überprüfte die Vorhersagegenauigkeit von CGformer in zwei Phasen: „Vortraining“ und „Feinabstimmung“.
Während der Vortrainingsphase (siehe Abbildung unten) zeigte CGformer eine überlegene Stabilität und Vorhersagegenauigkeit. Sein anfänglicher Fehler und seine Schwankung waren deutlich geringer als die von CGCNN. Eine zehnfache Kreuzvalidierung (10-facher CV) ergab, dass der mittlere absolute Fehler (MAE) des Trainingsdatensatzes 0,1703 betrug, was einer Verbesserung von 25,71 TP³T gegenüber CGCNN entspricht; der durchschnittliche MAE des Testdatensatzes betrug 0,3205, was einer Verbesserung von fast 101 TP³T gegenüber CGCNN entspricht. Vergleiche mit ALIGNN und SchNet unterstrichen die überlegene Leistung von CGformer zusätzlich.
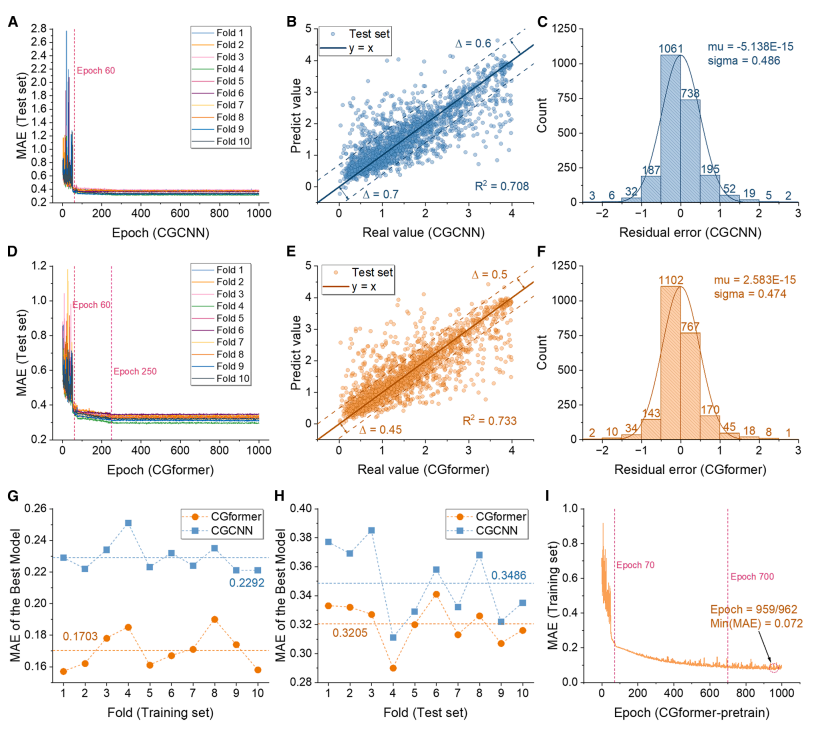
Aus den Anpassungsergebnissen geht hervor, dass der vorhergesagte Wert von CGformer weniger vom wahren Wert abweicht, der Rest stärker in der Nähe von 0 konzentriert ist und die Standardabweichung des Rests kleiner ist, was beweist, dass seine Vorhersage des Natriumions Eb zuverlässiger ist.
Während der Feinabstimmungsphase (wie in der Abbildung unten gezeigt) sank der MAE des vortrainierten CGformers nach etwa 10 von 200 Feinabstimmungsrunden deutlich, und der endgültige durchschnittliche MAE der 10-fachen Kreuzvalidierung betrug nur 0,0361. Nach der Feinabstimmung wurde die Abweichung zwischen dem vorhergesagten Wert und dem wahren Wert weiter reduziert, und die Residuen konzentrierten sich hauptsächlich im Bereich von -0,05 bis 0,05 und zeigten eine gute Normalverteilung, was seine extrem hohe Genauigkeit bei der Vorhersage des Hochentropiesystems Eb demonstriert und sein Anwendungspotenzial in Szenarien mit fehlenden Daten widerspiegelt.
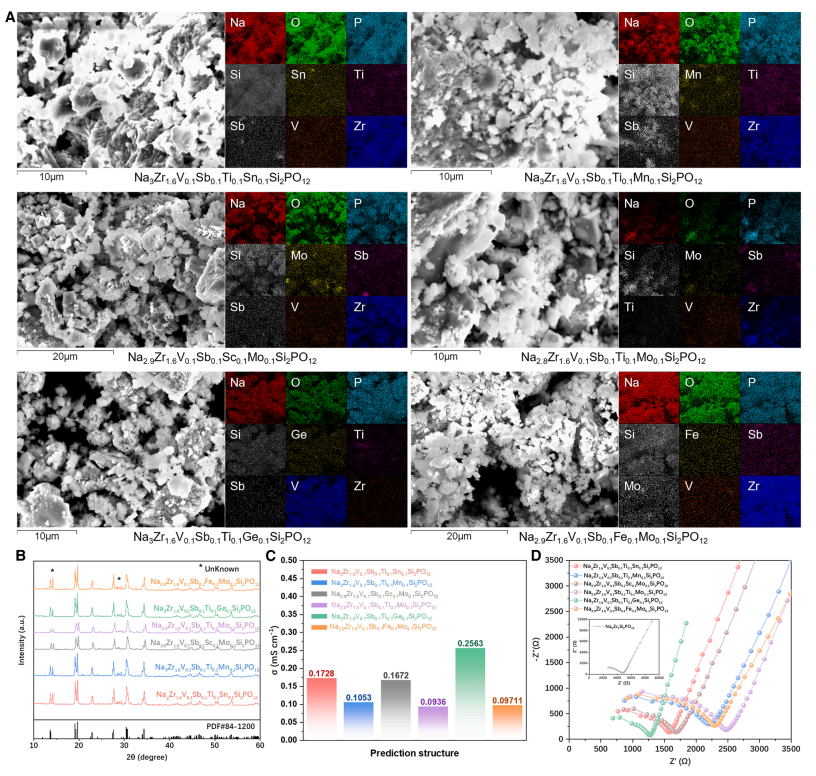
Abschließend wurden die sechs von CGformer ausgewählten optimalen HE-NSEs synthetisiert und elektrochemisch charakterisiert, um ihre Struktur und Leistung zu überprüfen. Die Ergebnisse zeigten, dass diese Materialien eine ausgezeichnete Ionenleitfähigkeit bei Raumtemperatur aufwiesen. Die Natriumionenleitfähigkeit lag bei 25 °C zwischen 0,093 und 0,256 mS/cm und damit deutlich höher als die von undotiertem Na₃Zr₂Si₂PO₁₂.
„Künstliche Intelligenz + Materialien“ ist zum Mainstream der Materialwissenschaftsentwicklung geworden
„Künstliche Intelligenz + Materialien“ hat sich zu einer hochmodernen Forschungsrichtung im aktuellen Bereich der Materialwissenschaften entwickelt. Durch die Integration von künstlicher Intelligenztechnologie mit Materialforschung und -entwicklung, Design und Anwendung wird das große Entwicklungspotenzial und der Anwendungswert der Schnittstelle zwischen den beiden Disziplinen deutlich. Die Einführung von CGformer hat zweifellos einen bedeutenden Beitrag zur Anwendung künstlicher Intelligenz im Bereich der Materialwissenschaften geleistet. Dank seiner einzigartigen und innovativen algorithmischen Architektur löst es die wichtigsten Probleme bei der Forschung und Entwicklung von Materialien mit hoher Entropie.
CGformer ist nur die Spitze des Eisbergs in der Erforschung der Bereiche künstliche Intelligenz und Materialwissenschaft durch das AIMS-Lab. Als eine der Hauptforschungsrichtungen des Labors ist die interdisziplinäre Forschung zu künstlicher Intelligenz und Materialwissenschaft seit langem zu einer lebendigen Fußnote des Labors geworden und hat fruchtbare Ergebnisse hervorgebracht.
Letztes Jahr präsentierte dasselbe Team seine Forschungsergebnisse in der führenden internationalen Fachzeitschrift Energy Storage Materials unter dem Titel „Transformer ermöglicht die Entwicklung des Ionentransportverhaltens und die Regulierung der Leitfähigkeit für Festelektrolyte“. Die Studie schlug ein künstliches Intelligenzmodell namens T-AIMD vor, das eine Transformer-Netzwerkarchitektur nutzt. Dieses Modell reduziert den Rechenaufwand erheblich und ermöglicht gleichzeitig eine schnelle und genaue Vorhersage des Verhaltens beliebiger Ionen in beliebigen Kristallstrukturen. Dieser Ansatz erhöht die Geschwindigkeit herkömmlicher AIMD-Simulationen um mehr als das Hundertfache und beschleunigt so die Bewertung von Materialeigenschaften erheblich.
Papieradresse:
https://www.sciencedirect.com/science/article/abs/pii/S2405829724003829
Unabhängig davon veröffentlichten Teams der Technischen Universität Berlin und der Universität Luxemburg in AIP Publishing eine entsprechende Forschungsarbeit und schlugen SchNet vor, eine Deep-Learning-Architektur, die speziell für die Simulation atomarer Systeme entwickelt wurde. Diese Architektur nutzt kontinuierlich filternde Faltungsschichten, um chemisch plausible Einbettungen von Atomtypen im Periodensystem zu erlernen und zeigt damit leistungsstarke Fähigkeiten zur Vorhersage verschiedener chemischer Eigenschaften von Molekülen und Materialien. Der Titel der Arbeit lautet „SchNet – Eine Deep-Learning-Architektur für Moleküle und Materialien“.
Papieradresse:
Von alltäglichen Kunststoffverpackungen und Metallprodukten bis hin zu Nanomaterialien und Supraleitern in High-End-Industrien ist der Fortschritt der menschlichen Zivilisation eng mit der Entwicklung der Materialwissenschaften verbunden. Die rasante Entwicklung der künstlichen Intelligenz wird zweifellos enormes Potenzial für die Zukunft der Materialwissenschaften freisetzen, was wiederum indirekt den Fortschritt der menschlichen Zivilisation vorantreiben wird.








