Command Palette
Search for a command to run...
Das MIT-Team Schlug Das FASTSOLV-Modell Vor, Das 50-mal Schneller Ist Als Das Ursprüngliche Modell, Um Die Löslichkeit Kleiner Moleküle Bei Jeder Temperatur vorherzusagen.

In der Chemie und Materialwissenschaft ist die Löslichkeit organischer Feststoffe in verschiedenen Lösungsmitteln eine zentrale Moleküleigenschaft, die sich auf die gesamte Forschungs- und Industriekette auswirkt. Bei Syntheseprozessen hilft die präzise Kontrolle der Löslichkeit nicht nur bei der Auswahl des optimalen Lösungsmittels und der Optimierung der Reaktionsbedingungen, sondern verbessert auch Produktausbeute und -reinheit deutlich und senkt so die Produktionskosten. In den Umweltwissenschaften ist sie ein Schlüsselparameter für die Analyse der Migration und des Verbleibs von Schadstoffen wie per- und polyfluorierten Alkylsubstanzen (PFAS) in Boden und Wasser und liefert eine wissenschaftliche Grundlage für die Vermeidung und Kontrolle von Umweltverschmutzung. Und bei Prozessen wie der Kristallisation und der Membrantrennung ist die Löslichkeit eine zentrale Variable, die das Phasenverhalten und die Trennleistung bestimmt.
Herkömmliche experimentelle Bestimmungsmethoden unterliegen jedoch zahlreichen Einschränkungen: Sie sind nicht nur zeit- und materialintensiv, sondern werden auch leicht durch Faktoren wie organische Feststoffkristalle und Verunreinigungen beeinträchtigt, was zu einer unzureichenden Datengenauigkeit führt. Untersuchungen zufolge beträgt die interlaboratorische Standardabweichung der Wasserlöslichkeit logS oft bis zu 0,5–0,7 log-Einheiten, und in Extremfällen kann die Differenz der Messergebnisse sogar das Zehnfache übersteigen. Obwohl empirische Gruppenadditionsmethoden, quantenchemische Modelle und Methoden des maschinellen Lernens zur Vorhersage eingesetzt wurden,Allerdings gibt es häufig Probleme mit unzureichender Vielseitigkeit oder Schwierigkeiten bei der Balance zwischen Genauigkeit und Rechenleistung.
Um dieses Problem zu lösen, kombinierte ein Forschungsteam des Massachusetts Institute of Technology chemische Informatiktools mit der neuen organischen Löslichkeitsdatenbank BigSolDB.Verbessert auf Basis der FASTPROP- und CHEMPROP-Modellarchitektur,Das Modell kann gleichzeitig gelöste Moleküle, Lösungsmittelmoleküle und Temperaturparameter eingeben und direkt ein Regressionstraining auf logS durchführen.
In einem strengen Solute-Extrapolationsszenario, verglichen mit bestehenden SOTA-Modellen wie Vermeire,Der RMSE des optimierten Modells wurde um das 2- bis 3-fache reduziert und die Inferenzgeschwindigkeit um das bis zu 50-fache erhöht.Derzeit hat das Team das FASTPROP-Derivatmodell FASTSOLV genannt und als Open Source veröffentlicht. Es bietet ein effizientes und praktisches Tool für die entsprechende wissenschaftliche Forschung und industrielle Anwendungen.
Die entsprechenden Forschungsergebnisse wurden in Nature Communication unter dem Titel „Data-driven organic solubility prediction at the limit of aleatoric uncertainty“ veröffentlicht.

Papieradresse:
https://www.nature.com/articles/s41467-025-62717-7
Folgen Sie dem offiziellen Konto und antworten Sie mit „Organic Solubility“, um das vollständige PDF zu erhalten
BigSolDB-gesteuerte Datensatzkonstruktion und Evaluierungssystemdesign
Die zentrale Datenquelle dieser Studie ist BigSolDB, das systematisch Löslichkeitsdaten organischer Feststoffe in einer Vielzahl organischer Lösungsmittel und unter verschiedenen Temperaturbedingungen nahe der Niederschlagsgrenze sammelt und so eine wichtige Unterstützung für das Training allgemeiner Vorhersagemodelle bietet.
Um das Forschungsziel zu erreichen, „neue gelöste Stoffe ohne jegliches Vorwissen zu extrapolieren“, entwickelte das Forschungsteam ein strenges Trainings- und Bewertungssystem:Das Modell wurde auf BigSolDB trainiert und unabhängig voneinander an zwei öffentlichen Datensätzen getestet: SolProp und Leeds.Um eine Unterschätzung der Schwierigkeit der Extrapolation zu vermeiden, wurden in dieser Studie, wie in der folgenden Abbildung gezeigt, zunächst alle gelösten Stoffe in SolProp entfernt, die sich mit BigSolDB überschnitten, und als Ergänzung der Leeds-Datensatz mit einem breiteren chemischen Raum eingeführt.
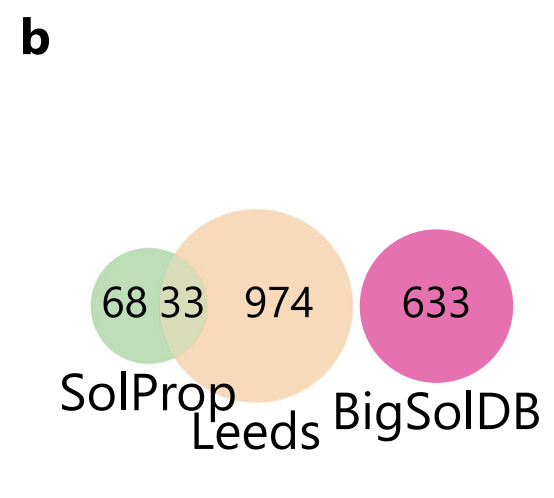
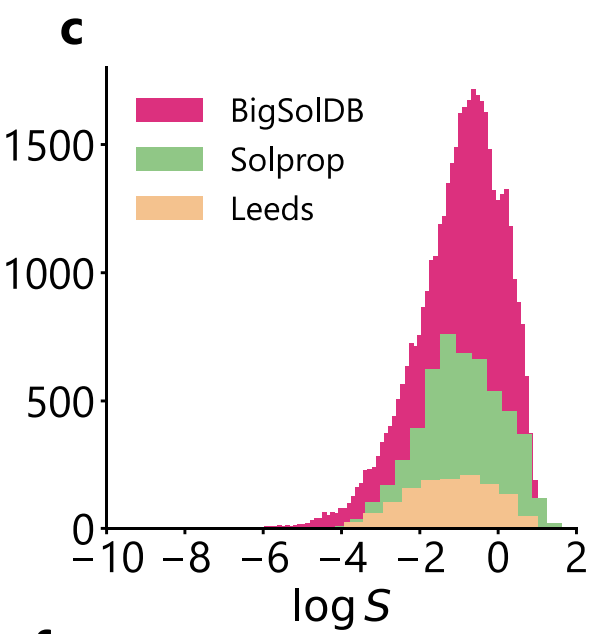
Im Vergleich zu SolProp,Leeds bietet eine höhere Vielfalt gelöster Stoffe, deckt jedoch nur die Bedingungen bei Raumtemperatur ab.Dies ermöglicht nicht nur das Testen der Anpassungsfähigkeit des Modells an neue chemische Räume, sondern bietet auch eine höhere obere Unsicherheitsgrenze aufgrund der fehlenden impliziten Rauschreduzierung durch „Mehrtemperaturmittelung“. Wie in der folgenden Abbildung gezeigt, sind die logS-Verteilungen für die drei Datensätze besonders konsistent, alle nahe –1 konzentriert und weisen einen langen Schwanz am Ende der niedrigen Löslichkeit auf, wodurch die Verteilungsvergleichbarkeit für Leistungsvergleiche zwischen den Datensätzen gewährleistet wird.
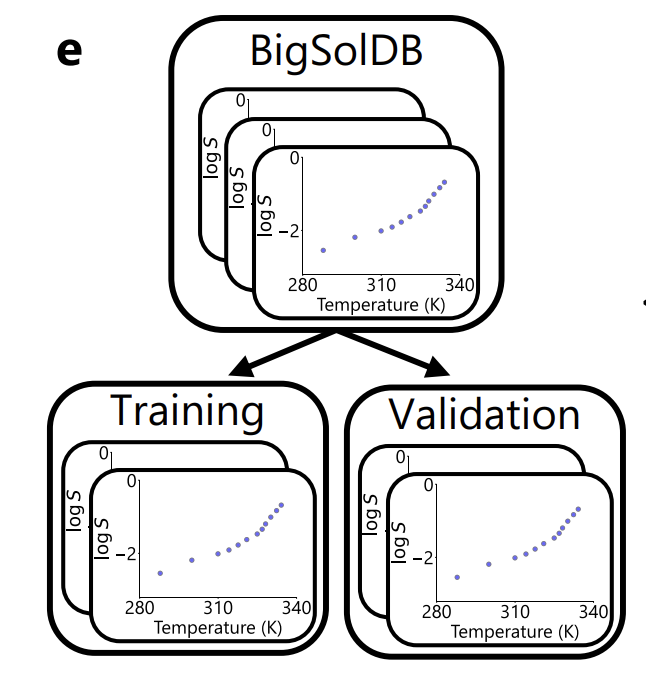
In Bezug auf die Datensegmentierung verwenden die Forscher, wie in der folgenden Abbildung gezeigt, ausschließlich gelöste Stoffe als Einheit: 95% des gelösten Stoffes werden für das Training verwendet, 5% werden für die Validierung und Modellauswahl verwendet,Alle Messungen desselben gelösten Stoffes in unterschiedlichen Lösungsmitteln und bei unterschiedlichen Temperaturen werden nicht gleichzeitig in unterschiedlichen Teilmengen angezeigt.Dadurch wird ein Informationsverlust wirksam vermieden.
Darüber hinaus wurde in der Studie das ASTARTES-Toolkit verwendet, um den Validierungssatz in den Trainingsdaten zufällig in „vollständige Experimente“ aufzuteilen, und die Aufteilungsgrenzen sowohl der gelösten als auch der experimentellen Dimensionen wurden in der abschließenden Auswertung erneut überprüft, um die Unabhängigkeit und Genauigkeit der Auswertung sicherzustellen.
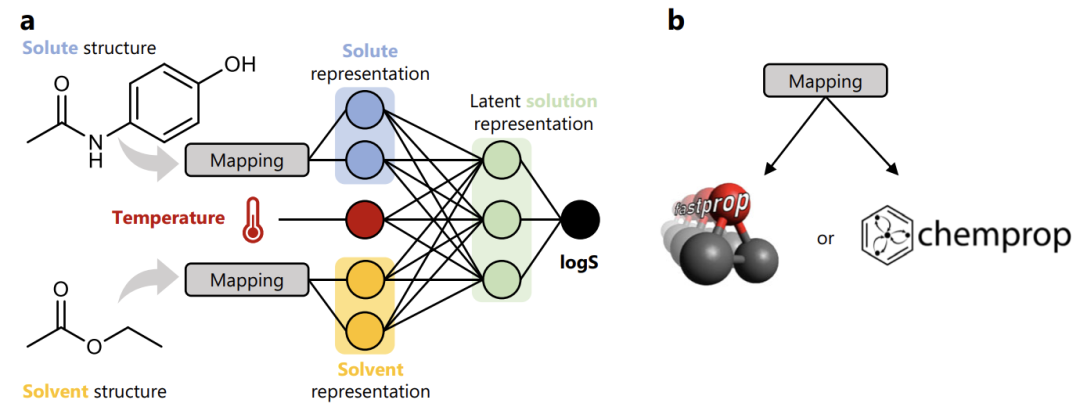
FASTSOLV-Modellkonstruktion gesteuert durch BigSolDB
Basierend auf dem BigSolDB-Datensatz, wie in der folgenden Abbildung dargestellt, hat diese Studie die beiden klassischen Modellarchitekturen von FASTPROP und CHEMPROP angepasst und einen klaren Modellierungsprozess für maschinelles Lernen erstellt.
Erste,Abbildung der Molekülstrukturen von gelösten Stoffen (z. B. Paracetamol) und Lösungsmitteln (z. B. Ethylacetat) in entsprechende Darstellungsvektoren;Dann,Diese beiden molekularen Darstellungsvektoren werden mit dem Lösungstemperaturparameter kombiniert, um eine vollständige Lösungsdarstellung zu bilden.Finale,Die Darstellung wurde in ein vollständig verbundenes neuronales Netzwerk eingegeben und ein Regressionstraining mit logS (Logarithmus der Löslichkeit) als Ziel durchgeführt.
Durch diese Transformation erreicht das schließlich entwickelte Modell eine einheitliche Vorhersage der Löslichkeit kleiner Moleküle in mehreren organischen Lösungsmitteln und verschiedenen Temperaturszenarien und überwindet damit die Abhängigkeit des traditionellen Modells von bestimmten Lösungsmitteln oder Temperaturbereichen.
Um die Robustheit und Vorhersagesicherheit des Modells weiter zu verbessern, verließ sich das Forschungsteam nicht auf eine einzelne Modellausgabe.Stattdessen wird das FASTPROP-Modell unter vier verschiedenen zufälligen Initialisierungsbedingungen trainiert und dann durch die Kombination der Integrationsstrategien das endgültige FASTSOLV-Modell erhalten.Alle nachfolgenden Schlüsselanalysen, wie Leistungsvergleiche und Fallüberprüfungen, basieren auf diesem integrierten Modell, wodurch das Risiko zufälliger Schwankungen eines einzelnen Modells effektiv reduziert wird.
Um die Leistung des neuen Modells objektiv zu messen, wurde in der Studie gleichzeitig das derzeit allgemein anerkannte SOTA-Modell, das Vermeire-Modell, als Vergleichsmaßstab eingeführt. Dieses Modell wird durch vier unabhängige thermochemische Untermodelle trainiert und gibt dann Löslichkeitsergebnisse durch Kombination thermodynamischer Zyklen aus. Es hat den Vorteil, Lösungsmittelvielfalt und Temperaturabhängigkeit auszugleichen. Die Studie ergab jedoch, dass der für seine Tests verwendete SolProp-Datensatz eine große Überschneidung der gelösten Struktur mit seinem eigenen Trainingssatz aufweist. Diese „Datenüberschneidung“ kann zu einer Überschätzung der extrapolierten Leistung führen. Um Fairness und Genauigkeit beim Vergleich zu gewährleisten, wurde in dieser Studie der ursprüngliche Trainings- und Testaufbau des Vermeire-Modells genau reproduziert und auf dieser Grundlage Kontrollexperimente durchgeführt, um sicherzustellen, dass der Leistungsunterschied nur auf das Modell selbst und nicht auf die Testbedingungen zurückzuführen ist.
Aktualisiert das SOTA für die Extrapolation der organischen Löslichkeit mit 2–3-facher Genauigkeit und 50-facher Geschwindigkeit
In dieser Studie wurden mehrdimensionale Tests durchgeführt und die Modellleistung überprüft. Im Interpolationsszenario erreichte das optimierte FASTPROP-Modell RMSE=0,22, P₁=94% und das CHEMPROP-Modell RMSE=0,28, P₁=90%.Die Leistung hat sich der Obergrenze des experimentellen Datenrauschens genähert und bestätigt den unterstützenden Wert von BigSolDB.
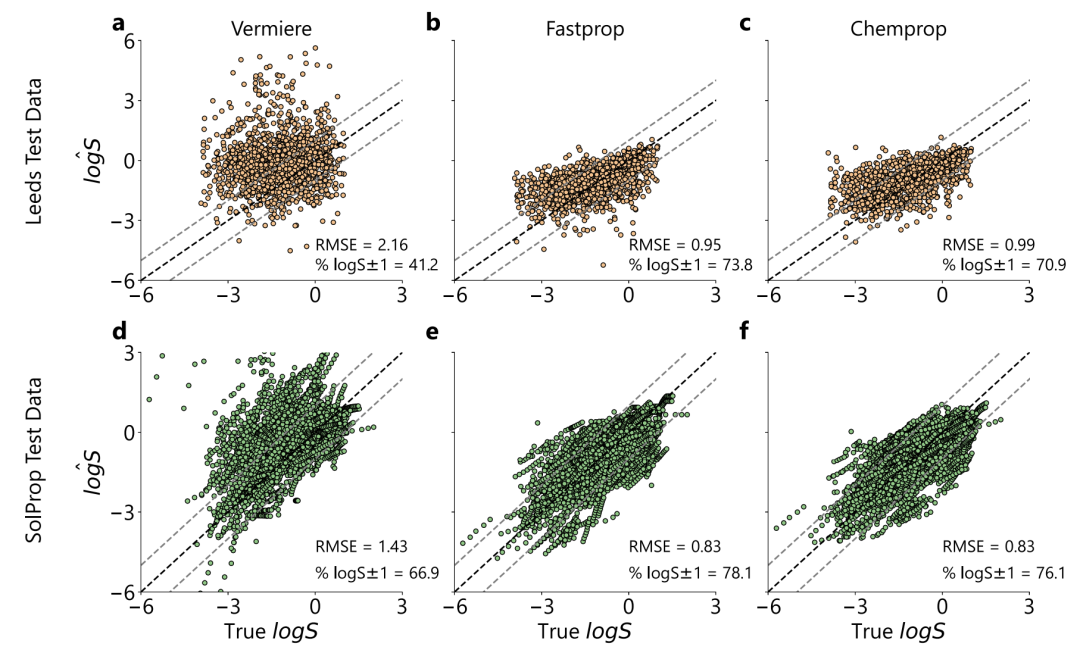
Im neuen Solute-Extrapolationstest, wie in der folgenden Abbildung dargestellt, schnitt das Vermeire-Modell im Leeds-Datensatz aufgrund systematischer Überschätzung schlecht ab (RMSE=2,16, P₁=34%), während der RMSE von FASTPROP und CHEMPROP auf 0,95 bzw. 0,99 sank und P₁ 69% überschritt. Im SolProp-Datensatz schnitt unser Modell ebenfalls besser ab (RMSE=0,83, P₁=80%).Und die Inferenzgeschwindigkeit von FASTPROP ist etwa 50-mal so hoch wie die des Vermeire-Modells.Unterstützt die SHAP-Interpretierbarkeitsanalyse.
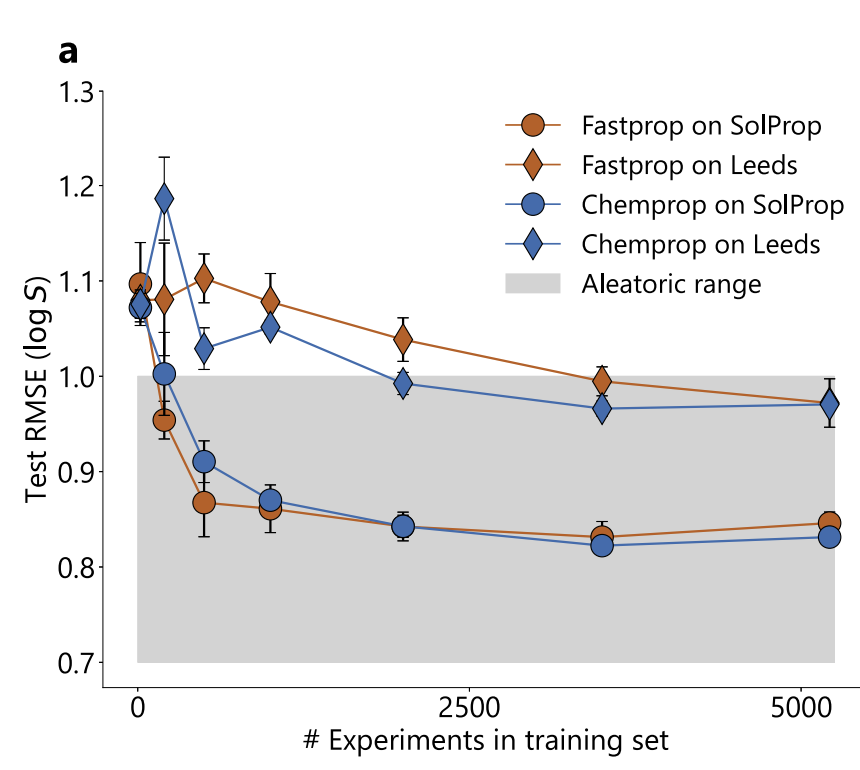
Das Experiment zum Trainingsdatenvolumen ist in der folgenden Abbildung dargestellt. Obwohl FASTPROP und CHEMPROP unterschiedliche Moleküldarstellungen aufweisen, konvergieren ihre Leistungen zu ähnlichen Grenzen: Der SolProp-Testsatz benötigt etwa 500 Experimente (≈5.000 Datenpunkte), um das Plateau zu erreichen, während CHEMPROP im Leeds-Testsatz etwa 2.000 Experimente (≈20.000 Datenpunkte) benötigt.
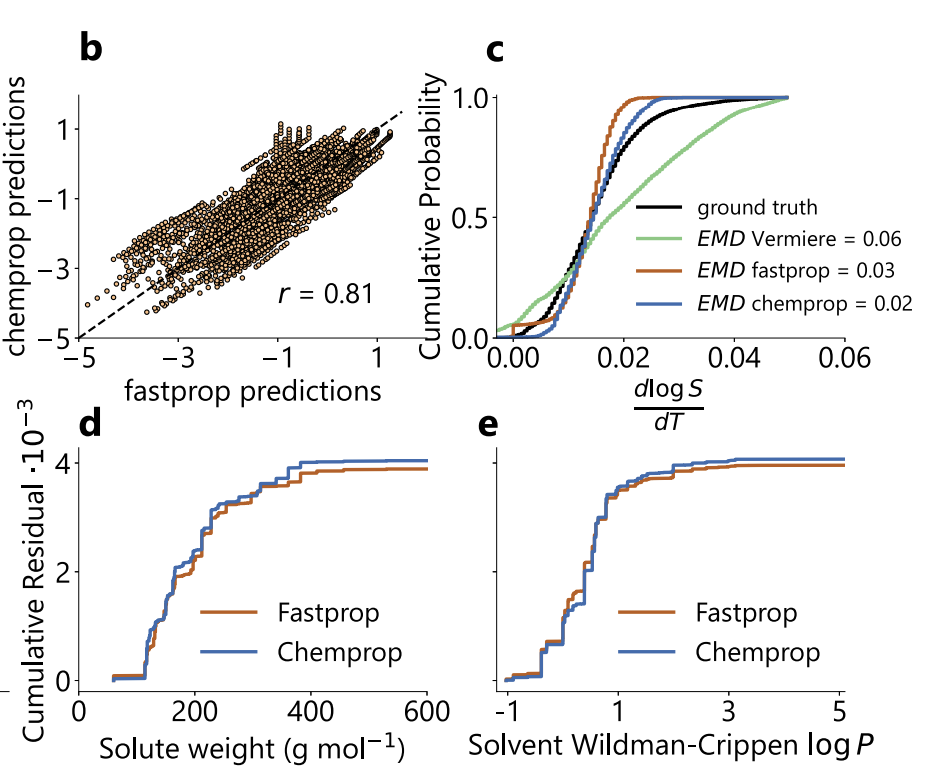
Geschätzt aus 34 Sätzen von Daten aus mehreren Quellen unter denselben Bedingungen in BigSolDB beträgt die experimentelle zufällige Unsicherheitsgrenze RMSE = 0,75 Log-Einheiten, während der RMSE der beiden Modelle auf SolProp 0,83 beträgt, was nahe an dieser Grenze liegt; im Vergleich zu großen Modellen wie MolFormer und ChemBERTa-2 schneiden die beiden Modelle besser ab.Dies beweist, dass der Leistungsengpass eher auf experimentelle Daten als auf die Ausdruckskraft des Modells zurückzuführen ist.
Mittelwerttest der Modellleistung bei beliebigen Grenzen
Darüber hinaus weisen die beiden Modelle, wie in der folgenden Abbildung dargestellt, im SolProp-Testsatz eine hohe Korrelation zwischen ihren Vorhersagen auf (Pearson r = 0,81), und auch die vorhergesagten Temperaturgradientenverteilungen sind sehr konsistent (EMD = 0,03/0,02). Der systematische Fehler ist deutlich geringer als beim Vermeire-Modell (EMD = 0,06).
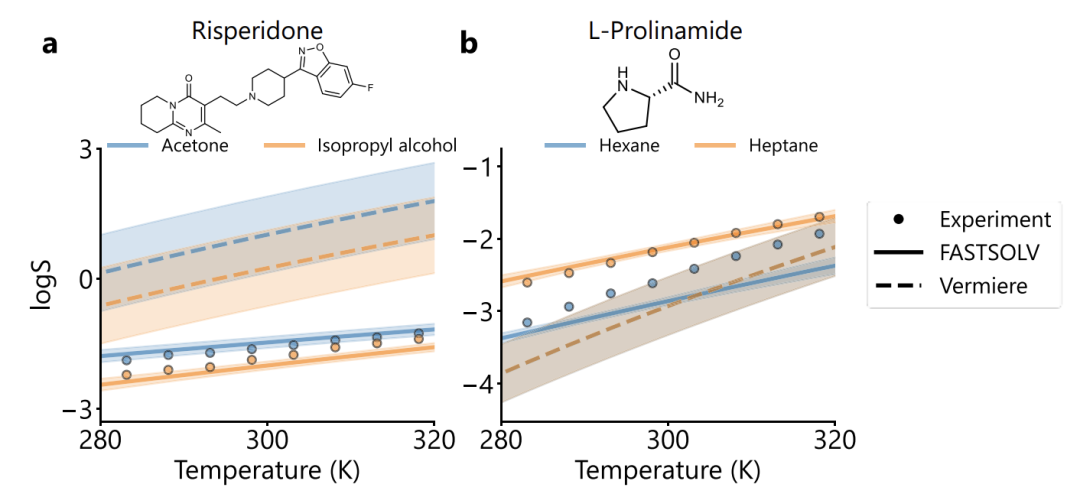
Die Studie ergab außerdem, dass FASTSOLV bei der typischen Solutevalidierung, wie in der folgenden Abbildung dargestellt, einen erheblichen Vorteil bei der Vorhersage von Risperidon (RMSE = 0,16 gegenüber Vermeire 1,64) und L-Prolin (RMSE = 0,25 gegenüber Vermeire 2,33) hat.Es kann nicht nur die Ordnungs- und Temperaturabhängigkeit der Lösungsmittellöslichkeit korrekt bestimmen, sondern auch zwischen Hexan und Heptan unterscheiden, die ähnliche Strukturen aufweisen.Die Fehlermodusanalyse zeigte, dass der Vorhersagefehler von Anthrachinonen hoch war, aber in der Teilmenge von 85 Anthrachinon/Anthrachinon-Derivaten betrug der Gesamt-RMSE des Modells 0,52 und die Lösungsmittellöslichkeit konnte stabil eingestuft werden, was darauf hindeutet, dass die molekulare Charakterisierung angemessen war.
Zusammenfassend:Im Vergleich zum Vermeire-Modell reduziert FASTSOLV den RMSE um das 2- bis 3-fache und beschleunigt die Inferenz um das bis zu 50-fache.Diese Methode vereint Interpretierbarkeit mit technischem Potenzial und stellt unter strengen Extrapolationseinstellungen eine hochmoderne Leistung dar. Die Studie weist auch darauf hin, dass durch das Hinzufügen zusätzlicher Trainingsdaten die Leistungsgrenzen nicht überschritten werden. Die zukünftige Forschung wird sich auf den Aufbau eines hochpräzisen Datensatzes für organische Lösungsmittel konzentrieren.
„Datensatz + KI“ ermöglicht weltweiten Durchbruch bei der Vorhersage molekularer Eigenschaften
In der heutigen Welle bereichsübergreifender Innovationen in Chemie, Medizin und Materialwissenschaften wird die auf „groß angelegten Datensätzen + fortschrittlichen Modellen des maschinellen Lernens“ basierende Technologie zur Vorhersage molekularer Eigenschaften zu einem wichtigen Instrument zur Bewältigung von Schwachstellen in der Branche, wie etwa zeitaufwändigen Experimenten, hohen F&E-Kosten und schwieriger Leistungsvorhersage.
Im akademischen Bereich reagieren Forschungsteams weltweit auf die Durchbrüche von FASTSOLV und BigSolDB mit der Einführung einer Reihe innovativer Studien zur Löslichkeitsvorhersage. So schlugen Forscher der britischen Universität Leeds beispielsweise ein Kausal-Struktur-Eigenschafts-Beziehungsmodell vor, das künstliche Intelligenz mit physikalisch-chemischen Mechanismen kombiniert.Die Löslichkeitsvorhersage in organischen Lösungsmitteln und Wassersystemen ist fast so genau wie der experimentelle Fehler.Darüber hinaus ist es hervorragend interpretierbar und gilt als wichtiger Meilenstein auf dem Gebiet der Löslichkeitsmodellierung.
Unterdessen hat ein Forscherteam am Massachusetts Institute of Technology (MIT) mithilfe des Graph-Neural-Network Chemprop bedeutende Fortschritte bei der Antibiotika-Forschung erzielt. Sie ermittelten die antibiotische Aktivität und die menschlichen Zytotoxizitätsprofile von 39.312 Verbindungen und nutzten Graph-Neural-Network-Ensembles, um die antibiotische Aktivität und Zytotoxizität von 12.076.365 Verbindungen für die Entwicklung neuer Antibiotika vorherzusagen. Durch das Screening einer Reihe von Ausgangsverbindungen und die Bewertung ihrer wachstumshemmenden Wirkung gegen den Methicillin-empfindlichen Stamm S. aureus RN4220,Es wurden 512 Wirkstoffe erhalten.Anschließend wird das Graph-Neuralnetzwerk trainiert, um binäre Klassifizierungsvorhersagen durchzuführen.
Auch in der Pharmaindustrie zeichnen sich bemerkenswerte Innovationen ab. Sie konzentriert sich seit langem auf kostengünstige Technologien zur Löslichkeitsbestimmung mit hohem Durchsatz. Beispielsweise kann das Tool Aspen Solubility Modeler von AspenTech die Löslichkeit in Hunderten von Lösungsmittelkombinationen auf Basis von Messdaten in wenigen Lösungsmitteln vorhersagen. Dieses Tool verbessert die Effizienz und Entscheidungssicherheit beim Kristall-Screening und der Prozessentwicklung bei großen Unternehmen wie GSK und AstraZeneca erheblich.
Darüber hinaus nutzen einige Unternehmen ähnliche datenbasierte Modelle in der Materialforschung und -entwicklung. Durch die Analyse großer Mengen an Molekülstruktur- und Leistungsdaten können sie die Eigenschaften neuer Materialien vorhersagen, F&E-Zyklen verkürzen und F&E-Kosten senken. In der chemischen Industrie verwenden einige Unternehmen Modelle, um die Auswirkungen chemischer Reaktionen unter verschiedenen Lösungsmittel- und Temperaturbedingungen vorherzusagen, Produktionsprozesse zu optimieren und die Produktionseffizienz sowie Produktqualität zu verbessern. Dies sind alles Beispiele dafür, wie Unternehmen Modelle und Datenkonzepte aus der akademischen Forschung auf tatsächliche Produktionsinnovationen anwenden.
Referenzlinks:
2.https://www.manufacturingchemist.com/news/article_page/Solubility_modelling/57726
Erhalten Sie mit einem Klick hochwertige Papiere und ausführliche Interpretationsartikel im Bereich AI4S von 2023 bis 2024 ⬇️









