Command Palette
Search for a command to run...
Eine Im Fachjournal „Nature“ Veröffentlichte Epidemiologische Abwasserbewertung Auf Grundlage Von Gensequenzierung Und Maschinellem Lernen Kann Viren Bis Zu Vier Wochen Früher erkennen.

In den letzten Jahren stand die globale öffentliche Gesundheitssicherheit vor großen Herausforderungen. Dies gilt insbesondere seit dem Ausbruch der COVID-19-Pandemie. Der Erreger, das Schwere Akute Respiratorische Syndrom Coronavirus 2 (SARS-CoV-2), hat sich weiterentwickelt, und es sind mehrere dominante Varianten entstanden. Diese Varianten verfügen über unterschiedliche Fähigkeiten, Infektionen auszulösen und Immunreaktionen zu umgehen, was die Prävention und Kontrolle der Epidemie erheblich erschwert und die Gesundheitssysteme zusätzlich belastet.
Tests im öffentlichen Gesundheitswesen und die Sequenzierung des SARS-CoV-2-Genoms sind wichtige Mittel zur umfassenden Erkennung zirkulierender Varianten.Allerdings ist diese Art der klinischen Überwachung oft stark von zahlreichen Laborressourcen abhängig und erfordert die aktive Teilnahme einzelner Personen an den Tests.Es ist schwierig, das Auftreten und die Verbreitung von SARS-CoV-2-Varianten vollständig zu verfolgen. Insbesondere in Gebieten mit relativ begrenzten medizinischen Ressourcen oder geringer Testbereitschaft ist die klinische Überwachung anfälliger für Erkennungsfehler, was zu blinden Flecken bei Prävention und Kontrolle führt.
Als ergänzender Ansatz spielt die Abwasserepidemiologie (WBE) eine wichtige Rolle bei der Warnung vor Krankheitsausbrüchen, seit sie in den 1940er Jahren erstmals zur Beurteilung von Infektionen in der Bevölkerung vorgeschlagen wurde. WBE erkennt und verfolgt die Zusammensetzung und dynamische Veränderungen von Viren hauptsächlich durch die Analyse von Spuren von Viren, die vom menschlichen Körper im Abwasser ausgeschieden werden.Im Vergleich zur klinischen Überwachung kann WBE die Gruppeninfektionssituation im abgedeckten Bereich objektiv und unvoreingenommen widerspiegeln, ohne auf individuelle aktive Tests angewiesen zu sein, eine Frühwarnung zu erreichen und weist eine erhebliche Kosteneffizienz auf.
Allerdings sind die derzeit gängigen Methoden zur Abwasserüberwachung (wie Freyja und COJAC auf Basis linearer Regression) noch immer mit Einschränkungen behaftet.Die Erkennung muss auf dem Mutationsmuster bekannter Varianten basieren (z. B. Referenzsequenzen in den Datenbanken GISAID oder UshER).Wenn eine neue Variante auftritt, die nicht charakterisiert oder in der klinischen Literatur aufgeführt ist, ist es oft schwierig, sie genau zu identifizieren, was die Erkennungseffizienz von WBE bis zu einem gewissen Grad einschränkt.
Um dieses Problem zu lösen, schlug ein Forschungsteam der University of Nevada, Las Vegas, eine multivariate Analysemethode namens ICA-Var (Independent Component Analysis of Variants) vor.Die Methode basiert auf einem unüberwachten Prozessdesign für maschinelles Lernen und verwendet die unabhängige Komponentenanalyse (ICA), um Kovariation und sich im Laufe der Zeit entwickelnde Mutationsmuster aus Abwasserdaten zu extrahieren.Dadurch wird eine frühere und genauere Variantenerkennung erreicht.
Mithilfe dieser Methode konnte das Forschungsteam zwischen Ende 2021 und 2023 die Delta-Variante, die Omicron-Variante und die rekombinante XBB-Variante präzise nachweisen. Diese Methode bestätigt nicht nur die Wirksamkeit der Abwasserüberwachung zur Frühwarnung bei der Prävention und Kontrolle von Epidemien, sondern bietet auch ein neues Instrument zur umfassenden Verfolgung viraler Mutationen und deren Ausbreitung ohne klinische Überwachung.
Die entsprechende Forschungsarbeit wurde in Nature Communications unter dem Titel „Früherkennung neu auftretender SARS-CoV-2-Varianten im Abwasser durch Genomsequenzierung und maschinelles Lernen“ veröffentlicht.
Forschungshighlights:
* Diese Methode deckt die räumlich-zeitliche Dynamik viraler Mutationen in städtischen und ländlichen Gebieten auf, bestätigt das Gesetz der Virusübertragung von städtischen in ländliche Gebiete und bietet ein effektives und kostengünstiges Paradigma zur Variantenerkennung für Gebiete mit schlechtem medizinischen Zugang oder Mangel an klinischen Sequenzierungsdaten
* Im Vergleich zum aktuellen Goldstandard-Tool Freyja bietet die multivariate Analysemethode von ICA-Var erhebliche Vorteile, und die Erkennungszeit von Delta, Omicron und den neuesten Varianten EG.5, HV.1, BA.2.86 ist durchschnittlich 1-4 Wochen früher

Papieradresse:
https://www.nature.com/articles/s41467-025-61280-5
Langfristige, mehrpunktige Datenerfassung
In dieser Studie wurden die im Experiment verwendeten Abwasserproben von August 2021 bis November 2023 gesammelt.3.659 Abwasserproben wurden aus städtischen und ländlichen Gebieten im Süden Nevadas gesammelt.Nach der Entnahme werden die Abwasserproben vor Ort auf Eis gelegt und bis zur Verarbeitung gekühlt aufbewahrt, die Lagerzeit beträgt höchstens 36 Stunden.
Während des NukleinsäureextraktionsprozessesDas Forschungsteam isolierte zunächst Nukleinsäuren aus Abwasserproben gemäß den gesetzlichen Vorschriften mit dem Promega Wizard Enviro Total Nucleic Kit (Kat.-Nr. A2991). Anschließend modifizierten sie das Promega-Protokoll, lysierten das Abwasser mit einer Proteaselösung und verwendeten Macherey-Nagel NucleoMag Beads (Kat.-Nr. 744970), um freie Nukleinsäuren zu binden. Für RNA über 10 ng verwendete das Team das New England BioLabs LunaScript RT SuperMix Kit für die Erststrang-cDNA-Synthese.
Aufbau und Sequenzierung von Sequenzierungsbibliotheken,Das Forschungsteam verwendete das CleanPlex SARS-CoV-2 FLEX Panel von Paragon Genomics, um Amplikon-Sequenzierungsbibliotheken zu erstellen, die dann auf der Illumina NextSeq 500- oder NextSeq 1000-Plattform mithilfe einer 300-Zyklen-Durchflusszelle sequenziert wurden.
Im Hinblick auf die Sequenzierung der DatenverarbeitungDas Team verwendete zunächst die Software cutadapt (Version 4.2), um Illumina-Adaptersequenzen aus Sequenzierungs-Read-Paaren zu entfernen. Anschließend ordneten sie die Sequenzierungs-Read-Paare mithilfe der Software bwa mem (Version 0.7.17-r1188) dem SARS-CoV-2-Referenzgenom (NC_045512.2) zu. Anschließend verwendeten sie das Tool fgbio TrimPrimers (Version 2.1.0, Hard-Trimming-Modus), um Paragon Genomics CleanPlex SARS-CoV-2 FLEX-Amplikon-Primersequenzen aus den ausgerichteten Reads zu entfernen. Schließlich wurde die Software iVar variants (Version 1.4.1) verwendet, um Varianten zu erkennen (basierend auf Allelfrequenzunterschieden im Vergleich zum ursprünglichen Referenzgenom von 2020), und die Software samtools (Version 1.16.1) wurde verwendet, um die Genomabdeckung und die Read-Tiefe zu berechnen.
Nach dem Entfernen doppelter Proben und positiver/negativer KontrollenDie restlichen 2.684 Proben wurden für die Qualitätskontrollanalyse (QC) verwendet.Nach einer strengen Qualitätskontrolle wurden für die anschließende Analyse nur Abwasserproben mit einer Sequenzierungstiefe von 50x und einer Abdeckung von mindestens 80% des SARS-CoV-2-Genoms aufbewahrt.
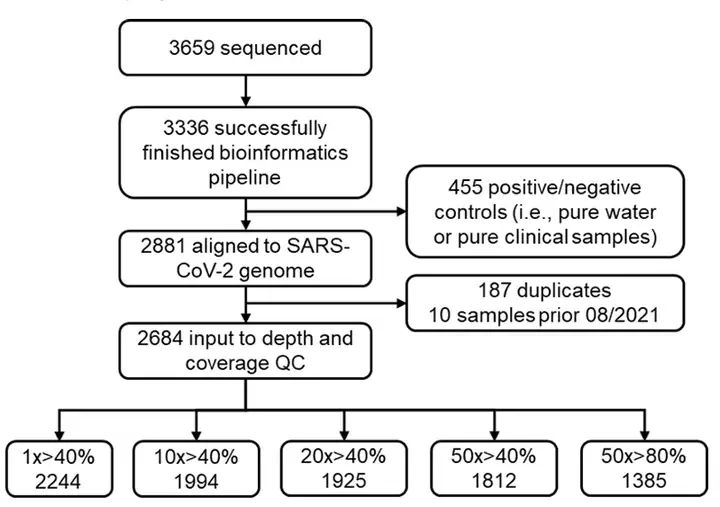
Finale,Für die Studie wurden 1.385 hochwertige Proben verwendet.Deckt 59.422 Mutationsstellen von SARS-CoV-2-Varianten für die anschließende Analyse ab.
Um die Wirksamkeit der ICA-Var-Methode zu überprüfen, verwendete das Forschungsteam klinische Daten als Kontroll- und Referenzbasis und analysierte 8.810 klinische SARS-CoV-2-Sequenzdaten mit hoher Abdeckung aus Nevada, die aus der GISAID-Datenbank heruntergeladen wurden und den Zeitraum von September 2021 bis November 2023 abdecken.
Mit ICA als Kern wird eine duale Regressionsmethode eingeführt, um ein neues Tool zur COVID-19-Erkennung zu schaffen
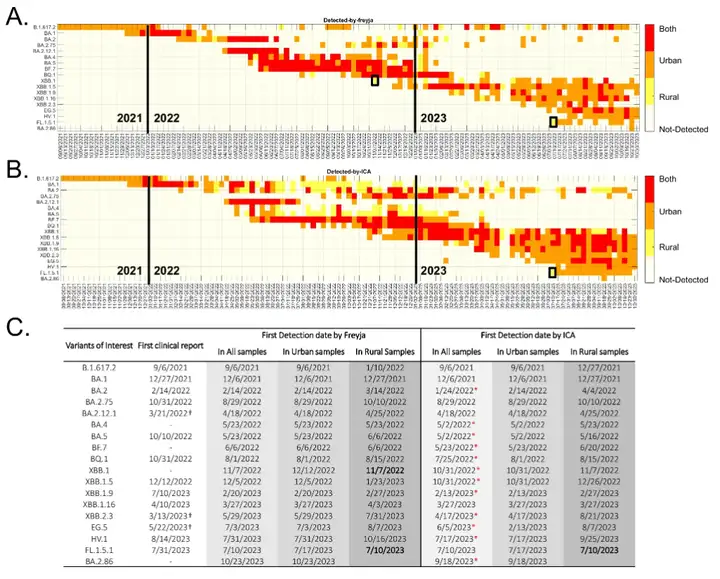
Der Kernprozess von ICA-Var istEs verarbeitet die Mutationshäufigkeiten in Abwasserproben durch unabhängige Komponentenanalyse und extrahiert unabhängige Kovariationsmutationsmuster.Diese Muster werden dann durch duale Regression mit den Originalproben verknüpft, um Virusvarianten zu verfolgen, wie in der folgenden Abbildung dargestellt:
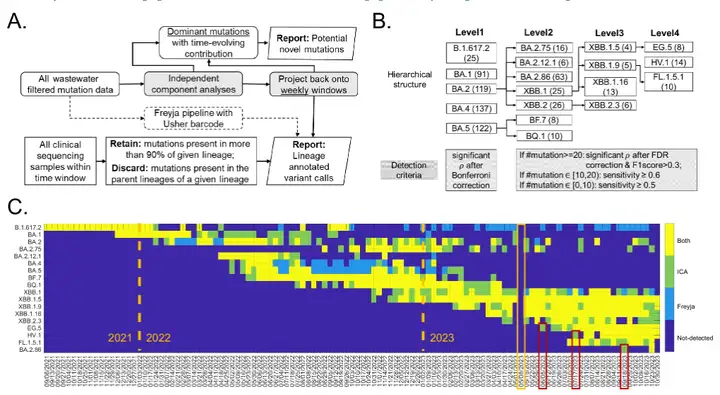
* Abbildung A ist der unabhängige Komponentenanalyseprozess.Die beiden Matrizen sind: wöchentliche SARS-CoV-2-Linienerkennung (untere Reihe) und potenzielle neue Mutationen (obere Reihe)
* Abbildung B zeigt die hierarchische Struktur von 18 besorgniserregenden Varianten.Die wichtigsten Mutationsstellen jeder Variante (d. h. die liniendefinierenden Stellen) wurden entnommen aus http://covspectrum.org Zusammengefasste klinische Daten mit der Anzahl der Hauptmutationen in Klammern und den schattierten Kästchen, die die im vorgeschlagenen Arbeitsablauf zu testenden Kriterien angeben.
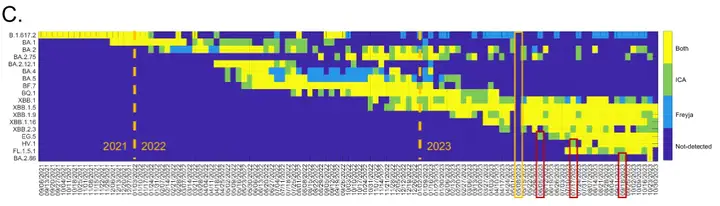
*Abbildung C zeigt den Vergleich zwischen der ICA-Var-Methode und dem hochmodernen Tool Freyja.Bei den neu aufgetretenen Varianten EG.5, HV.1 und BA.2.86 zeigt das rote Kästchen einen früheren ICA-Var-Erkennungszeitpunkt an; das gelbe Kästchen zeigt eine Woche an, in der aufgrund technischer Probleme keine Abwasserproben entnommen wurden.
Da das SARS-CoV-2-Genomsignal in Abwasserproben das Ergebnis einer Mischung mehrerer Varianten ist und durch Probenzersetzung, Sequenzierungsfehler usw. gestört wird, ist es mit herkömmlichen Methoden schwierig, die Eigenschaften einer einzelnen Variante direkt zu analysieren.Die Kernidee von ICA-Var besteht in der Verwendung einer unabhängigen Komponentenanalyse——Diese Technologie zur blinden Quellentrennung geht davon aus, dass das gemischte Mutationssignal eine lineare Kombination mehrerer „unabhängiger Quellen“ ist, und verwendet mathematische Modellierung, um diese unabhängigen Muster von den gemischten Daten zu trennen.
Das Forschungsteam hat die Daten zunächst vorverarbeitet.Durch die Qualitätskontrolle der SARS-CoV-2-Genomsequenzierungsdaten aus Abwasserproben, bei der minderwertige Messwerte und verrauschte Mutationen herausgefiltert wurden, wurde eine „Mutationshäufigkeitsmatrix“ erstellt, deren Zeilen die Proben, Spalten die Mutationsstellen und Werte die Mutationshäufigkeit jeder Stelle in der Probe darstellen. Anschließend wurde eine unabhängige Komponentenanalyse der Mutationshäufigkeitsmatrix durchgeführt, bei der das gemischte Signal in unabhängige Komponenten zerlegt wurde. Jede Komponente repräsentiert einen Satz von „Kovariationsmutationsmustern“ oder Kombinationen von Mutationen, die für eine bestimmte Variante charakteristisch sind und im Laufe der Zeit synchron in allen Proben auftreten oder verschwinden.
Hier,In der Studie wurde das Kriterium der minimalen Beschreibungslänge (MDL) verwendet, um die Anzahl der unabhängigen Komponenten zu bestimmen, und eine unabhängige Komponentenzerlegung mithilfe des fastICA-Algorithmus durchgeführt.Um die Zuverlässigkeit der Ergebnisse sicherzustellen, wiederholten sie die ICA-Analyse 50 Mal mit unterschiedlichen Anfangswerten, gruppierten und visualisierten die in jedem Durchlauf erhaltenen Komponenten mithilfe der ICASSO-Software und behielten schließlich nur die zuverlässigen Schätzungen bei, die den engen Clustern als Quellmatrix entsprachen.
Anschließend, um die wöchentliche Variantensituation weiter zu bestimmen,Das Forschungsteam verwendete die Methode der doppelten Regression, um die aus der unabhängigen Komponentenanalyse gewonnene Quellmatrix auf die ursprüngliche Stichprobe zu projizieren.Berechnen Sie den „Beitrag“ jeder unabhängigen Komponente in jeder Probe, d. h. die relative Häufigkeit der Variante in der Probe, um die dynamischen Veränderungen verschiedener Varianten in Zeit und Raum zu quantifizieren, wie z. B. den Zeitpunkt des Auftretens, epidemische Trends und Unterschiede in der Verteilung zwischen Stadt und Land.
Das Forschungsteam verwendete die Vollproben-Quellmatrix als Satz von Quellregressoren in einem allgemeinen linearen Modell (GLM), um die Signalzerlegungsmuster für jede wöchentliche Stichprobe zu ermitteln, die mit der Vollproben-Quellmatrix in Zusammenhang steht. Anschließend nutzten sie die Signalzerlegungsmuster für jede wöchentliche Stichprobe als Regressoren in einem zweiten GLM, um die wochenspezifische Quellmatrix zu ermitteln, die weiterhin mit der Vollproben-Quellmatrix in Zusammenhang steht. Dieser Prozess erzeugte Paare von Schätzungen, die den dualen Raum bildeten und zusammen die beste Annäherung an die ursprüngliche Vollproben-Quellmatrix der unabhängigen Komponentenanalyse in jeder wöchentlichen Stichprobe lieferten.
endlich,Das Forschungsteam verglich die isolierten unabhängigen Komponenten mit bekannten Varianten in klinischen Sequenzierungsdaten und annotierte sie.Dadurch kann der entsprechende Variantenstamm erfolgreich bestimmt oder nicht übereinstimmende Kovariationsmutationsmuster herausgefiltert werden, um vor der Möglichkeit neuer Variantenstämme zu warnen.
Die ICA-Var-Methode überwindet die Nachteile herkömmlicher Methoden, die auf „vordefinierten Referenzvarianten-Barcodes“ beruhen.Durch die Erfassung der Kovariationsmuster von Mutationen ist es möglich, neue Varianten früher und genauer zu identifizieren als mit herkömmlichen Methoden.In Kombination mit einer dualen Regressionsanalyse zeigt diese Methode auch Unterschiede bei der Übertragung in städtischen und ländlichen Gebieten sowie die zeitliche Entwicklung von Mutationsstellen auf. Zusammenfassend lässt sich sagen, dass ICA-Var ein sensitiveres, umfassenderes und kostengünstigeres Instrument zur COVID-19-Erkennung darstellt.
Die Erkennungseffizienz übertrifft das aktuelle Goldstandard-Tool Freyja und hat das Potenzial, neue Varianten vorherzusagen
Um die Leistung von ICA-Var zu validieren und zu bewerten, verglich das Forschungsteam es mit dem aktuellen Goldstandard-Tool Freyja, einem Tool zur Schätzung der relativen Häufigkeit von SARS-CoV-2-Linien im Abwasser. Es verwendet eine „Barcode“-Bibliothek aus Mutationen, die Linien definieren, um alle bekannten SARS-CoV-2-Linien eindeutig zu identifizieren, und verwendet eine tief gewichtete Regressionsmethode mit minimaler absoluter Abweichung, um die Linienhäufigkeit zu berechnen.Experimente haben bestätigt, dass die multivariate Analysemethode ICA-Var größere Vorteile bietet.
Wie in der folgenden Abbildung gezeigt, wurde im Abschnitt „Modellmethode und Architektur“ kurz erläutert, dass ICA-Var die neuen Varianten EG.5, HV.1 und BA.2.86 bereits früher erkennen kann. Der Hauptinhalt wird in diesem Abschnitt erweitert.
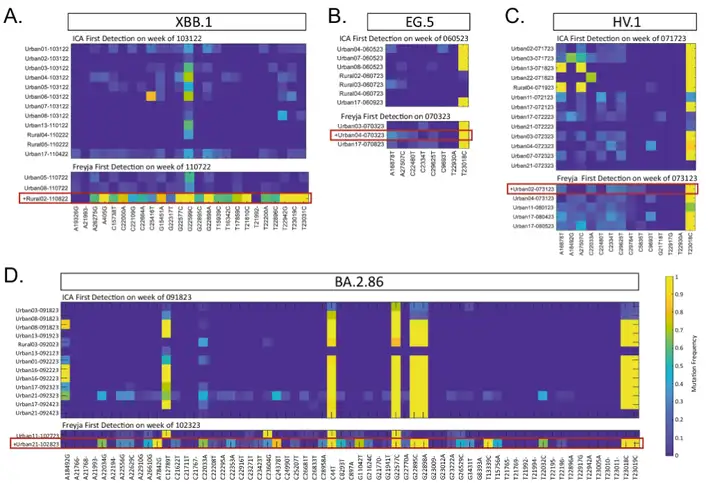
Konkret heißt das im Jahr 2022:Es wurde gezeigt, dass ICA-Var Varianten wie BA.2, BA.4, BA.5, BF.7, BQ.1, XBB.1 und XBB.1.5 eine oder mehrere Wochen früher erkennt als Freyja.Bei der Erkennung von EG.5 entdeckte ICA-Var diese Variante in der Woche vom 5. Juni, Freyja identifizierte das Signal von EG.5 jedoch erst am 3. Juli, als die Häufigkeit der Abwasserproben 23.08% erreichte und bereits fünf der acht dominanten Mutationsstellen von EG.5 angezeigt wurden. Ähnliches gilt für Varianten wie XBB.1, HV.1 und BA.2.86.ICA-Var wurde auch mehrere Wochen früher als Freyja entdeckt.
Dies liegt daran, dass ICA-Var Informationen aus mehreren Proben über „zuverlässige, aber geringe Prävalenz dominanter Mutationsstellen“ integriert., wodurch die statistische Aussagekraft verbessert und eine frühere Erkennung ermöglicht wird. Das bedeutet, dass es nicht auf einen hohen Anteil dominanter Mutationen in einer einzelnen Probe angewiesen ist; es kann die Erkennung einfach durch die Zusammenführung schwacher Signale aus mehreren Proben verbessern. Im Gegensatz dazu benötigt Freyja mindestens eine einzelne Probe, die eindeutig eine dominante Mutationsstelle aufweist, um die Erkennung abzuschließen. Das bedeutet auch, dass es stärker auf ein ausreichend starkes Mutationssignal in einer einzelnen Probe angewiesen ist und weniger empfindlich auf schwache oder verstreute Signale reagiert.
Das Experiment untersuchte außerdem die dynamischen Trends von Varianten in städtischen und ländlichen Proben. Ab Anfang 2022 sequenzierte und analysierte das Forschungsteam Abwasserproben aus ländlichen Gebieten im Süden Nevadas und führte einen umfassenden epidemiologischen Stadt-Land-Vergleich durch, bei dem städtische und ländliche Proben wöchentlich getrennt analysiert wurden.
Die Ergebnisse zeigten, dass sowohl ICA-Var als auch Freyja von den 18 besorgniserregenden Varianten zunächst 16 SARS-CoV-2-Varianten in städtischen Abwasserproben entdeckten, bevor sie in ländlichen Proben gefunden wurden. Dies deutet darauf hin, dass Virusvarianten in der Regel zuerst in Städten auftreten und sich dann auf ländliche Gebiete ausbreiten. Wie in der folgenden Abbildung dargestellt:
Die Ausnahme besteht darin, dass Freyja XBB.1 zunächst in ländlichen Abwasserproben entdeckte, während ICA-Var die Variante eine Woche zuvor in städtischen Abwasserproben entdeckte; beide Tools fanden FL.1.5.1 in ländlichen Abwasserproben, während die Häufigkeit und Prävalenz des alternativen Allels der dominanten Mutation dieser Variante in städtischen Abwasserproben im gleichen Zeitraum viel geringer war.
Die Studie enthüllte auch die zeitlichen Evolutionstrends von Mutationsstellen. Das Forschungsteam verglich 177 Mutationsstellen mit signifikanten zeitlichen Evolutionsbeiträgen zwischen August 2021 und November 2023 mit den dominanten Mutationsstellen der Varianten B.1.617.2, BA.1 und XBB.1, wie in der folgenden Abbildung dargestellt:
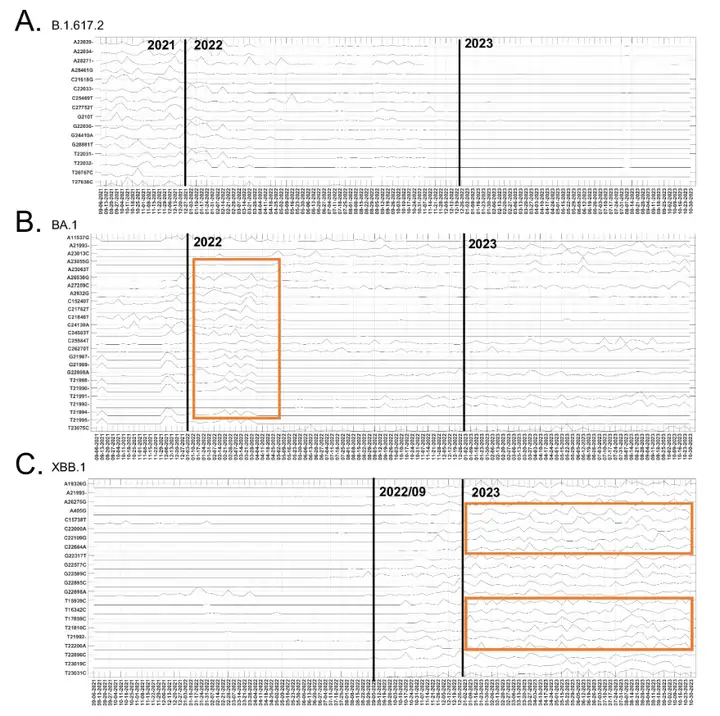
Von den 25 Hauptmutationsstellen in der Delta-Variante (B.1.617.2) zeigten 16 Ende 2021 signifikante Schwankungen im Beitrag, gefolgt von einem allmählichen Rückgang im Jahr 2022. Der Beitrag verwandter Mutationen im Omicron-Subtyp BA.1 stieg Ende 2021 signifikant an und erreichte Anfang 2022 seinen Höhepunkt. Der Beitrag einiger BA.1-Mutationsstellen schwankte 2023 weiterhin und wurde in anderen Omicron-Sublinien wie XBB.1 gefunden. Von den 25 Hauptmutationen in der XBB.1-Variante zeigten 22 signifikante zeitlich dynamische Beiträge mit signifikanten Auswirkungen nach September 2022. Mehrere Mutationsstellen zeigten ähnliche Fluktuationsmuster, was auf Kovariation hindeutet und die Rekombinationseigenschaften von XBB.1 widerspiegelt.
Diese Analysen zeigten, dass die zeitlichen evolutionären Beiträge der von ICA-Var identifizierten Mutationsstellen mit den klinischen Befunden der Varianten Delta, Omicron und XBB.1 übereinstimmten. Dies verdeutlichte die Zuverlässigkeit der ICA-Var-Ergebnisse und demonstrierte das Potenzial der Methode, neue Mutationsmuster zu identifizieren, die zur Entstehung neuer Varianten führen können.
Das Experiment überprüfte dies detailliert. Das Forschungsteam untersuchte 113 potenzielle neue Mutationsstellen, indem es sie mit den dominanten Mutationsstellen von 15 Hauptvarianten verglich. Anschließend nutzte es einen hierarchischen Clusteralgorithmus, um diese Mutationsstellen in sechs charakteristische Cluster zu klassifizieren. Wie in der folgenden Abbildung dargestellt:

Von diesen charakteristischen Clustern überschneiden sich die Mutationsstellen von vier (Cluster 2–5) mit den Ende 2023 aufgetretenen Varianten. Cluster 1 und Cluster 6 weisen keine überlappenden Mutationen mit bekannten Mutationsstellen auf. Die Mutationsstellen von Cluster 1 zeigten nach August 2023 ein deutliches Kovariationsmuster. Die klinischen Sequenzierungsdaten von GISAID zeigten, dass acht der Mutationsstellen verifiziert wurden und in klinischen Proben eine niedrige Meldehäufigkeit aufwiesen. DaherDiese Mutationen können zur Entstehung neuer Coronavirus-Varianten führen, die durch klinische Tests weiter verifiziert werden müssen.Eine genaue Überwachung ist erforderlich.
Die Abwasserüberwachung wird durch maschinelles Lernen weiterentwickelt, um eine hochwertige Virenprävention und -kontrolle zu ermöglichen.
Wie eingangs erwähnt, handelt es sich bei der WBE nicht um eine neue Methode. Bereits in den 1940er Jahren erkannten Umweltvirologen den Wert der Gewinnung von Polioviren durch Zellkulturexperimente im Abwasser. Seitdem wurde die WBE kontinuierlich verbessert und hat sich zu einem wirksamen Instrument zur Frühwarnung vor Krankheitsausbrüchen entwickelt.Seit dem Ausbruch der COVID-19-Pandemie hat WBE erneut eine positive Rolle bei der Prävention und Kontrolle der Epidemie gespielt.
Ende 2023 gab es beispielsweise Berichte, dass ein schwedisches Forschungsteam durch die Integration genomischer Tests von Abwasser und COVID-19-Fällen das Auftreten der neuen SARS-CoV-2-Variante BA.2.86 erfolgreich nachgewiesen habe. Um WBE effektiver und aktiver zur Erkennung neuer Coronavirus-Varianten nutzen zu können, haben viele Labore außerdem entsprechende Modelle entwickelt oder verbessert, um kostengünstigere Werkzeuge für WBE bereitzustellen.
So veröffentlichten Forscher der Tsinghua-Universität, der Hebei University of Science and Technology und des Tianjin Ecological and Environmental Monitoring Center gemeinsam eine Studie mit dem Titel „Validierung von Methoden zur Anreicherung und Erkennung von SRAS-CoV-2-RNA in Abwasser“. In der Studie wurden zwei Anreicherungstechniken – Ultrafiltration und kovalente Affinitätsharztrennung – mit zwei Nachweismethoden – der quantitativen Reverse-Transkriptase-PCR (RT-qPCR) und der digitalen Reverse-Transkriptase-PCR (RT-dPCR) – verglichen, um ihre Leistung bei der Überwachung von Abwasserviren zu bewerten.
endlich,Die Studie ergab, dass die Methode der digitalen Reverse-Transkriptase-PCR (RT-dPCR) die bessere Wahl für den Nachweis geringer Konzentrationen von SARS-CoV-2-RNA im Abwasser ist.Die Nachweisrate ist höher und die Toleranz gegenüber PCR-Inhibitoren ist besser.
* Papieradresse:
https://link.springer.com/article/10.1007/s10311-025-01843-6
Darüber hinaus veröffentlichte das Team um Professor Xingfang Li vom Department für Pathologie und Labormedizin der University of Alberta, Kanada, eine Studie mit dem Titel „Quantifizierung und Differenzierung von SARS-CoV-2-Varianten in Abwasser für Überwachungszwecke“. Basierend auf den zuvor für klinische Proben entwickelten Gamma- (ABG) und Delta-Multiplex-RT-qPCR-Nachweismethoden zielten sie auf die Omicron-Subvariante ab und nutzten ihre einzigartigen Mutationen.Es wurde ein Omicron-Triplex-RT-qPCR-Test entwickelt, der fünf Hauptunterlinien von Omicron-Varianten unterscheiden kann.Dies ist die erste Studie, bei der ein Single-Tube-RT-qPCR-Triplex-Assay verwendet wird, um alle Omicron-Subvarianten in Abwasserproben über einen Zeitraum von einem Jahr zu erkennen und zu identifizieren.
* Papieradresse:
https://pubs.acs.org/doi/10.1021/envhealth.3c00089
Kurz gesagt: Die Welt steht heute vor großen Herausforderungen für die öffentliche Gesundheit und Sicherheit. Dabei spielt die Abwasserüberwachung als hochwirksames Mittel zur Bevölkerungsüberwachung eine unersetzliche Rolle. Mit dem kontinuierlichen technologischen Fortschritt wird sich die Abwasserüberwachung weiterentwickeln – von der gezielten Früherkennung anhand bekannter Mutationsmuster bis hin zu Durchbrüchen bei der Sequenzierung des gesamten Genoms und der Identifizierung unbekannter Krankheitserreger. Mit zunehmender Sensibilität und Abdeckung liefert die Abwasserüberwachung präzisere und wichtigere Daten für die Epidemiewarnung, -verfolgung und Politikgestaltung und wird so zu einer unverzichtbaren Ergänzung der öffentlichen Gesundheit und Sicherheit.
Quellen:
1.https://www.nature.com/articles/s41467-025-61280-5
2.https://mp.weixin.qq.com/s/ZzzZt-uNNc5DsD-ib3Ww8g








