Command Palette
Search for a command to run...
Basierend Auf 86.000 Proteinstrukturdaten Wurden Mithilfe Einer Methode Des Maschinellen Lernens in Kombination Mit Quantenmechanischen Berechnungen 69 Neue Stickstoff-Sauerstoff-Schwefel-Bindungen Entdeckt

In der Zellfabrik fungiert die Stickstoff-Sauerstoff-Schwefel-Bindung (NOS) als reversibler „intelligenter Schalter“, der die Enzymaktivität entsprechend den Redoxänderungen in der Umgebung regulieren kann. Im Jahr 2021 entdeckte ein Team der Georg-August-Universität Göttingen durch die Untersuchung der Transaldolase von Neisseria gonorrhoeae die NOS-Bindung zwischen Lysin und Cystein.Diese Forschung geht über den Rahmen einzelner Pathogene und Enzymstudien hinaus und legt einen wichtigen Grundstein für die interdisziplinäre Proteinwissenschaft, Arzneimittelentwicklung und Biotechnik.
Mit dem explosionsartigen Wachstum der Daten über Proteinstrukturen und der fortgesetzten Erforschung der chemischen Bindungen in Proteinstrukturen sind jedoch auch neue Probleme entstanden.Gibt es andere NOS-Bindungen oder chemische Wechselwirkungen, die übersehen wurden?
Basierend auf den obigen Überlegungen,Sophia Bazzi und Sharareh Sayyad von der George Augustus University haben einen innovativen Algorithmus für die Computerbiologie entwickelt: SimplifiedBondfinder.Dies eröffnet ein neues Kapitel in der Erforschung kovalenter Proteinbindungen.Das Team integrierte maschinelles Lernen und quantenmechanische Berechnungen, um eine hochauflösende Röntgenkristallographie-Datenbank aufzubauen, und analysierte systematisch mehr als 86.000 hochauflösende Röntgenproteinstrukturen.Es wurden nicht nur 69 neue NOS-Bindungen entdeckt, sondern darunter auch neuartige NOS-Bindungen zwischen Arginin (Arg)-Cystein und Glycin (Gly)-Cystein, die noch nie zuvor beobachtet worden waren.
Diese revolutionäre Entdeckung erweiterte den Anwendungsbereich der Proteinchemie und ermöglichte eine gezielte Regulierung bei der Arzneimittelentwicklung und Proteintechnik.Obwohl sich diese Studie auf die NOS-Bindung konzentrierte, kann der Ansatz flexibel angewendet werden, um eine breite Palette anderer chemischer Bindungen und kovalenter Modifikationen zu untersuchen.Beinhaltet strukturell auflösbare posttranslationale Modifikationen (PTMs).
Die Forschungsergebnisse wurden in Communications Chemistry unter dem Titel „Revealing arginine-cysteine and glycine-cysteine NOS linkages by a systematic re-evaluation of protein structures“ veröffentlicht.
Forschungshighlights:
* Unter Aufhebung der gängigen wissenschaftlichen Annahme, dass NOS-Bindungen nur zwischen Lysin (Lys) und Cystein existieren, wurde der neue Redox-Regulationsmechanismus von Arginin-Cystein- und Glycin-Cystein-NOS-Bindungen erstmals mit einer innovativen Methode aufgedeckt
* Die vorgeschlagene Methode integriert maschinelles Lernen, quantenmechanische Berechnungen und hochauflösende Röntgenkristallographiedaten. Sie löst das Problem des Mangels an systematischen Algorithmen zur Entdeckung chemischer Bindungen in diesem Forschungsbereich, löst sich von den Beschränkungen traditioneller Experimente und bietet ein zuverlässiges und einfach zu bedienendes Werkzeug für die nachfolgende Forschung.
* Durch maschinelles Lernen und künstliche Intelligenz konnten die Kosten dieser Forschung deutlich gesenkt und gleichzeitig die Forschungseffizienz verbessert werden. Dies ist ein Beispiel für maschinenlernbasierte Technologien bei der Entschlüsselung von Proteinfunktionen und der Identifizierung neuer Proteininteraktionen.

Papieradresse:
https://www.nature.com/articles/s42004-025-01535-w
Weitere Artikel zu den Grenzen der KI:
https://go.hyper.ai/UuE1o
Datensatz: Extrahieren zuverlässiger Datensätze mit mehreren Einschränkungsebenen
Die von SimplifiedBondfinder gesammelten Daten stammen aus drei verschiedenen Proteindatenbanken.Es handelt sich um PDB, PDB-REDO und BDB. Die gesammelten Daten unterliegen verschiedenen Einschränkungen, um zuverlässige und nutzbare Datensätze herauszufiltern. Die Datenbank PDB-REDO (Stand: Januar 2024) verfeinert und optimiert die statische Struktur der PDB, um sie besser an aktuelle kristallographische Standards anzupassen. Im Vergleich zum ursprünglichen PDB-Eintrag weist sie eine höhere Genauigkeit und Zuverlässigkeit auf. Wie die linke Seite der folgenden Abbildung zeigt:
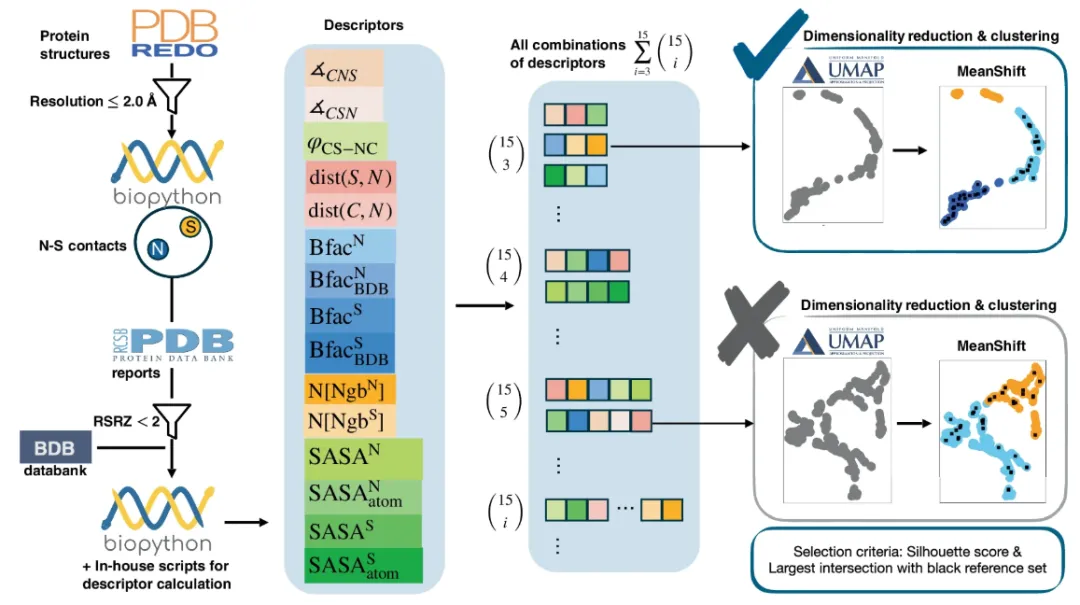
Insbesondere nutzte das Forschungsteam mehrere miteinander verbundene Funktionen, um die automatische Datensatzgenerierung in einer Datenbank voranzutreiben, die ursprünglich 170.251 Proteindaten enthielt.Zunächst wurde Biopython (Version 1.79) verwendet, um Strukturanalysen (mit MMCIFParser und PDBParse) durchzuführen und weitere Atom- und Resteigenschaften zu berechnen. Nach der Analyse der durch Röntgenstrahlen ermittelten Struktur optimierte das Forschungsteam die Daten von 170.127 Proteinen.
Um die Vorhersagegenauigkeit weiter zu verbessern, untersuchte das Forschungsteam anschließend weitere Proteinstrukturen mit einer Auflösung von ≤ 2 Å und erhielt schließlich 86.491 Strukturen für die experimentelle Analyse.
Um einen Datensatz für die Untersuchung einer bestimmten chemischen Bindung zu erstellen,Das Forschungsteam legte Kriterien auf Grundlage der Atomtypen, der Namen der Reste, der interatomaren Abstände und der Besetzung fest.Für NOS-Verbindungen mit Schwefel- (S) und Stickstoffatomen (N) in Standardresten beschränkte das Forschungsteam den interatomaren Abstand von SN, d. h. dist(S, N), auf ≤ 3,2 Å, entsprechend dem Grenzwert für Valenzwechselwirkungen zwischen Lysin und Cystein. Die Besetzungsschwelle wurde auf > 0,8 festgelegt, um Atome mit hoher Positionsunsicherheit auszuschließen. Anhand dieses Kriteriums identifizierte die Studie 25.462 NS-Kontakte.
Um sicherzustellen, dass die Zielatommasse dargestellt wird, hat das Forschungsteam zusätzlich den Realraum-R-Wert-Z-Score (RSRZ) mit einem Schwellenwert von <2,0 angewendet, um sicherzustellen, dass zuverlässige Übereinstimmungen mit den Daten im Realraum identifiziert werden konnten.Der Datensatz wurde weiter auf 23.129 NS-Kontakte reduziert.Dadurch konnten sich die experimentellen Ziele hauptsächlich auf zwei Arten von Wechselwirkungen von Cystein konzentrieren: die Wechselwirkung zwischen dem Schwefelatom von Cystein und dem Rückgratstickstoff von Glycin; und die Wechselwirkung zwischen dem Schwefelatom von Cystein und dem Seitenkettenstickstoff von Arginin und Lysin.
Nächste,Das Forschungsteam verwendete das NeighborSearch-Modul in Biopython, um Strukturparameter zu extrahieren und sammelte 15 verschiedene Deskriptoren für jede Probe in jedem Datensatz.Dazu gehören Winkel (∡CSN, ∡CNS), Torsionswinkel (φCS-NC), andere Entfernungen (dist(C, N), dist(S, N)) und die Werte der lösungsmittelzugänglichen Oberfläche (SASA) der Zielatome und der entsprechenden Reste, die weiter mit Bio.PDB.SASA berechnet werden.
Das Forschungsteam berücksichtigte atomare B-Faktoren (Bfac) im Experiment, um einen Zielparameter für die atomare Mobilität in der Analyse zu haben. Diese Werte stammten aus zwei Datenbanken, der RCSB PDB und einer PDB-Dateidatenbank (BDB) mit konsistenten B-Faktoren.
Es ist erwähnenswert, dass aufgrund der spezifischen Anforderungen dieser Studie im Experiment nur 15 Deskriptoren ausgewählt wurden.Das Forschungsteam erklärte jedoch, dass es für den vorgeschlagenen Algorithmus keine strikte Begrenzung hinsichtlich der Anzahl der Deskriptoren gebe, die er verarbeiten könne.Aufgrund seiner Konzeption kann es eine beliebige Anzahl von Deskriptoren aufnehmen, was die Integration domänenspezifischen Wissens oder die Anpassung an neue experimentelle Ansätze ermöglicht.
Modellarchitektur: Integration von maschinellem Lernen und Quantenmechanik-Computing
Der obige Teil ist der erste Schritt in den Schlüsselschritten der vorgeschlagenen Methode, die darin besteht, einen Zieldatensatz für bestimmte chemische Bindungen zu erstellen und strenge Kriterien anzuwenden.Dieser Abschnitt konzentriert sich auf den zweiten wichtigen Schritt der vorgeschlagenen Methode, nämlich die Verwendung von Techniken des maschinellen Lernens zur Untersuchung dieser hochdimensionalen Daten.Identifizieren Sie effektive Strukturdeskriptoren und sagen Sie potenzielle Stellen für die Bildung kovalenter Bindungen voraus.
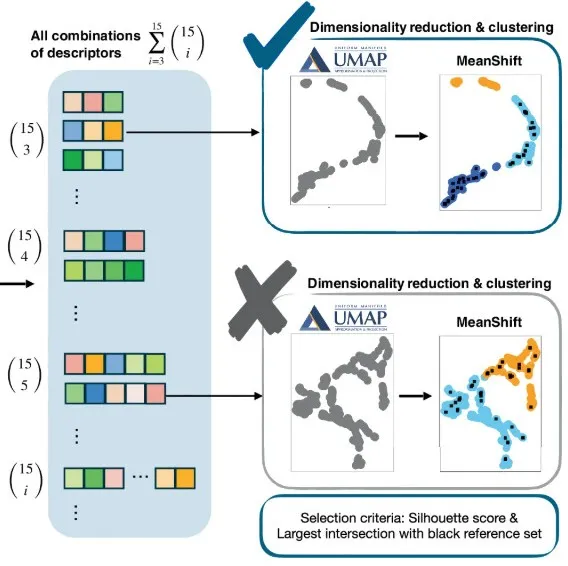
Wie im Bild oben gezeigt.Zunächst wandte das Forschungsteam die unbeaufsichtigte Dimensionsreduktionstechnik Uniform Manifold Approximation and Projection (UMAP) mit einer maximalen Einbettungsdimension von 3 an.Anschließend wird für alle möglichen Deskriptorensätze ein Mean-Shift-Clustering durchgeführt.
In,UMAP bewahrt die intrinsischen topologischen und geometrischen Eigenschaften hochdimensionaler Daten optimal und stellt sicher, dass wesentliche Strukturmerkmale in niedrigdimensionalen Einbettungen erhalten bleiben.Dies erleichtert eine aussagekräftige nachfolgende Analyse. Die Wahl der Einbettungsdimension in UMAP hängt von den topologischen und geometrischen Eigenschaften des Datensatzes und seiner ursprünglichen hochdimensionalen Mannigfaltigkeit ab. In praktischen Anwendungen sind zwei- oder dreidimensionale Einbettungen am besten interpretierbar, da sie eine intuitive Visualisierung und Bewertung der Clusterqualität ermöglichen.
In dieser Studie lieferten drei Einbettungsdimensionen gut getrennte und aussagekräftige Cluster, was die Wahl rechtfertigte. Die Ergebnisse der chemischen Bindungsanalyse und der Clusterbildung zeigen, dass diese Methode der Dimensionsreduzierung für den in diesem Experiment verwendeten Datensatz optimal ist. Die Wahl höherer Einbettungsdimensionen als nötig kann die ursprünglichen beliebten Merkmale erhalten, erhöht jedoch den Rechenaufwand, ohne die Interpretierbarkeit zu verbessern. Umgekehrt führt eine Reduzierung der Dimensionalität unter das optimale Niveau zu einem hohen Informationsverlust und einer schlechten Clustertrennung.
Dann,Das Forschungsteam ermittelte den Silhouette Score aller dreidimensionalen Einbettungskoordinaten, um die Clusterqualität jeder Kombination zu bewerten.Der Algorithmus gibt Cluster, Silhouettenkoeffizienten und die Referenz-Ziel-Konnektivität innerhalb jedes Clusters aus. Jeder Kandidat wird durch den Namen des Zielatoms, den entsprechenden Restnamen, die Restnummer, die Kette und die PDB-ID identifiziert, um alle Zielatome innerhalb des Proteins zu unterscheiden.
Um den endgültigen und minimalen Merkmalsraum zu finden, verwendete das Forschungsteam mehrere Kriterien, darunter den Wert des Silhouettenkoeffizienten, die Anzahl der von jedem Merkmalsraum erzeugten Cluster und die Verteilung der Referenz-Ziel-Verbindungen in diesen Clustern.
Speziell,Ziel des Forschungsteams war es, einen Merkmalsraum zu identifizieren, der die Daten effektiv in zwei oder drei unterschiedliche Cluster mit einem Silhouettenkoeffizienten ≥ 0,5 segmentieren kann.Idealerweise enthält einer der Cluster keine Referenzzielverbindungen. Dieser Cluster wird als „unmöglicher Cluster“ bezeichnet. In der Praxis ist die Mindestanzahl an Referenzproben in diesem Cluster akzeptabel. Die verbleibenden Cluster, die alle oder die meisten Referenzzielverbindungen enthalten, werden als „mögliche Cluster“ bezeichnet.
Durch die Einführung möglicher und unmöglicher Kandidatencluster, die die gewünschten chemischen Bindungen enthalten,Dem Forschungsteam gelang es, optimierte Merkmalsräume zu identifizieren, um zwischen Zielatompaaren zu unterscheiden, die wahrscheinlich neue chemische Bindungen bilden, und solchen, bei denen dies weniger wahrscheinlich ist.Sobald ein Satz von Deskriptoren identifiziert ist, der diese Fälle zuverlässig unterscheiden kann, ist die Einbeziehung zusätzlicher Deskriptoren nicht mehr erforderlich. Dieser Ansatz bietet Vorteile sowohl hinsichtlich der Rechenleistung als auch der Interpretierbarkeit und könnte die Vorhersagegenauigkeit von Methoden zur Identifizierung neuer chemischer Bindungen in Proteinstrukturen deutlich verbessern.
Zusätzlich zum maschinellen Lernen integriert die in dieser Studie vorgeschlagene Methode auch quantenmechanische Berechnungen.Die Forscher führten eine Geometrieoptimierung an potenziellen Kandidaten für die NOS-Verknüpfung in Lys-NOS-Cys-, Gly-NOS-Cys-, ARG-NηOS-Cys- und ARG-NεOS-Cys-Komplexen durch. Die Geometrieoptimierung erfolgte in Wasser mit dem Softwarepaket Gaussian16 – A.03 (Gaussian 16, Revision C.01) auf der Theorieebene B3LYP-D3 (BJ)/def2-TZVPD. Für die optimierten Strukturen wurden mehrere geometrische Parameter experimentell berechnet, darunter der Abstand zwischen Schwefel- und Stickstoffatomen (dist (S, N)) sowie die Winkel (∡CSN, ∡CNS, ∡NOS).
Um die Existenz von NOS-kovalenten Bindungen zu überprüfen, die durch die vorgeschlagene Clustermethode vorhergesagt wurden,Das Forschungsteam verwendete phenix.refine (Version 1.20.1-4487-000), um vier repräsentative Proteinstrukturen neu zu optimieren;Eine umfassende Strukturvalidierung wurde mit phenix.molprobity durchgeführt, um die geometrische Qualität, Kollisionswerte und sterischen Wechselwirkungen zu bewerten und die Übereinstimmung mit hochauflösenden kristallografischen Daten sicherzustellen. Ein vollständiger Validierungsbericht wurde mit phenix.table1 erstellt, der Verfeinerungsstatistiken, Modellqualitätsmetriken und stereochemische Abweichungen zusammenfasst. Diese Validierungsschritte bestätigten die strukturelle Integrität der NOS-Verbindung und ihre Kompatibilität mit der Elektronendichtekarte.
Experimentelle Ergebnisse: Arg-NOS-Cys und Gly-NOS-Cys Bindungen sind sinnvolle kovalente Bindungen
Um die Wirksamkeit der vorgeschlagenen Methode zu demonstrieren, führte das Forschungsteam eine Reihe von Experimenten durch und untersuchte dabei den Einsatz von Techniken des maschinellen Lernens für die Deskriptorenauswahl, die biochemische Bedeutung des Multideskriptorenraums, die Clusteranalyse sowie die strukturelle und thermodynamische Verifizierung.
Auswählen von Deskriptoren mithilfe von maschinellem Lernen
Das Forschungsteam wandte es zunächst auf Daten an, bei denen wahrscheinlich Lys-NOS-Cys-Verbindungen existierten.Der Datensatz enthält 527 Lysin-Cystein-Paare und beinhaltet auch experimentell verifizierte NOS-Bindungen.Als Schlüsseldeskriptoren wurden experimentell der B-Faktor des Stickstoffatoms (Bfac(BDB)(N)) und die Anzahl benachbarter Reste innerhalb eines Radius von 4 Å der Cα-Atome von Lysin (Ngbᴺ) und Cystein (Ngbˢ) ermittelt.
Das Forschungsteam erweiterte die Analyse weiter auf einen Datensatz von 313 Glycin-Cystein-Paaren, um potenzielle Gly-NOS-Cys-Verbindungen zu untersuchen, wie in der folgenden Abbildung dargestellt.
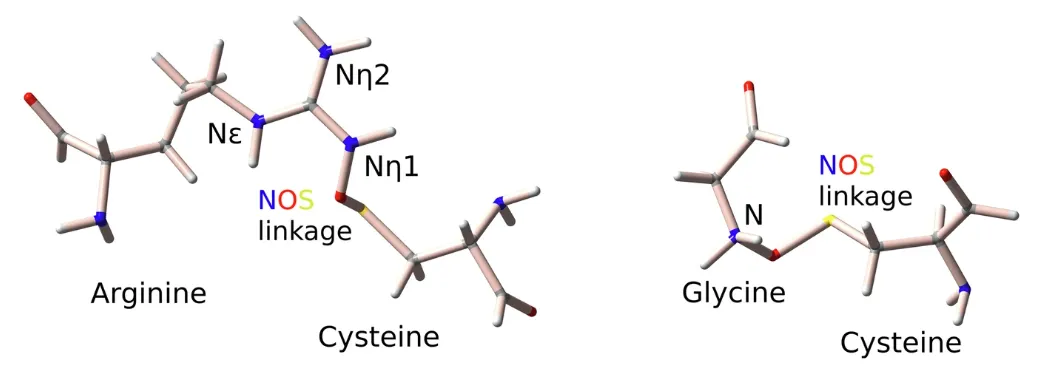
Zu den wichtigsten Deskriptorsätzen zählen hier der B-Faktor schwefelhaltiger Rückstände (BfacBDBS), der Schwefel-Stickstoff-Abstand (dist(S,N)) und der Kohlenstoff-Schwefel-Stickstoff-Winkel (∡CSN).
In Bezug auf Schlüsseldeskriptoren zur Vorhersage der NOS-Bindungsbildung zwischen Arginin- und Cysteinresten,Die Arginin-Seitenkette hat zwei Arten von Stickstoffatomen, Nη und Nε, die sich in ihren geometrischen Merkmalen und chemischen Eigenschaften unterscheiden.Daher haben wir die Datensätze Nη (Arg-NηOS-Cys) und Nε (Arg-Nε-Cys) separat analysiert.
Für Arg-NηOS-Cys entsprechen die ausgewählten Deskriptoren der für Lösungsmittel zugänglichen Oberfläche des Stickstoffrests (SASAᴺ), ∡CSN und den Resten neben Schwefel (Ngbˢ) und Stickstoff (Ngbᴺ); in ähnlicher Weise betreffen die Schlüsseldeskriptoren für den Datensatz von 240 Arg-NεOS-Cys-Paaren BfacBDBS, SASAˢ, die für Lösungsmittel zugängliche Oberfläche des Stickstoffatoms, ∡CSN und ∡CNS.
Diese Ergebnisse zeigen eine klare Clustertrennung durch die Visualisierung der UMAP-Dimensionsreduzierung.Wie in der folgenden Abbildung dargestellt, stellen Himmelblau und Königsblau Kandidaten für NOS-Bindungen dar, Orange steht für „unmögliche Clusterbildung“ und die schwarzen Quadrate stellen den Referenzdatensatz dar. Es ist deutlich zu erkennen, dass sich die Verteilung der Proben, die NOS-Bindungen bilden können, stark mit der Verteilung der Referenzstandardpunkte überschneidet.
Biochemische Bedeutung des multidimensionalen Deskriptorraums
Das Forschungsteam untersuchte die biochemische Relevanz von Schlüsseldeskriptoren, die für die Unterscheidung zwischen NOS- und Nicht-NOS-Bindungen wichtig waren, indem es algorithmisch einen Mindestsatz von Deskriptoren ermittelte.
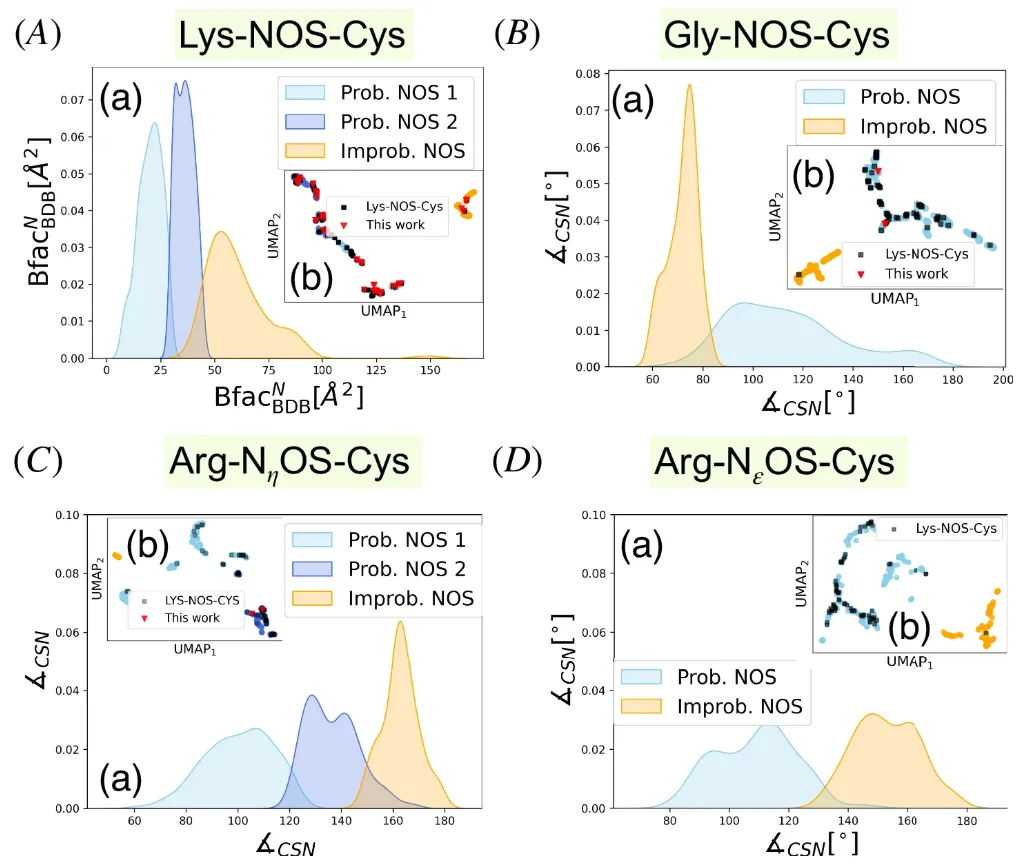
Am Beispiel des B-Faktors zeigt der B-Faktor in verschiedenen Clustern unterschiedliche Verteilungsmuster. Wie in A(a) oben gezeigt, sind die Modi des B-Faktors für „mögliches Clustering“ und „unmögliches Clustering“ unterschiedlich.Der B-Faktor hängt mit der Flexibilität von Atomen oder Regionen zusammen, und Reste im aktiven Zentrum haben normalerweise einen niedrigeren B-Faktor, was darauf hinweist, dass sie mit der Enzymaktivität zusammenhängen.Das Forschungsteam wies jedoch auch darauf hin, dass der niedrige B-Faktor möglicherweise auf eine NOS-Bindung hinweist, aber auch andere Stickstoff-Schwefel-Wechselwirkungen widerspiegeln könnte.
Hinsichtlich der Deskriptoreigenschaften von NOS-Bindungen, die durch verschiedene Aminosäurereste gebildet werden, ist BfacBDBᴺ der Hauptfaktor, der die beiden Cluster in Lys-NOS-Cys unterscheidet; bei Gly-NOS-Cys-Verbindungen ist ∠CSN der Hauptdeskriptor, der mögliche NOS-Verbindungscluster unterscheidet, wobei ∠CSN >80° für die meisten möglichen Proben ist und der ∠CSN-Wert des optimierten Gly-NOS-Cys-Komplexes ungefähr 94° beträgt; ∠CSN ist immer noch der entscheidende Faktor, um mögliche von unmöglichen NOS-Verbindungen für Arg-NεOS-Cys-Verbindungen zu unterscheiden.
Clusteranalyse
Bei dieser Auswertung entdeckte das Forscherteam 65 Lys-NOS-Cys-Bindungen, 2 Gly-NOS-Cys-Bindungen (Abbildungen a und b unten) und 2 Arg-NηOS-Cys-Bindungen (Abbildungen c und d unten).
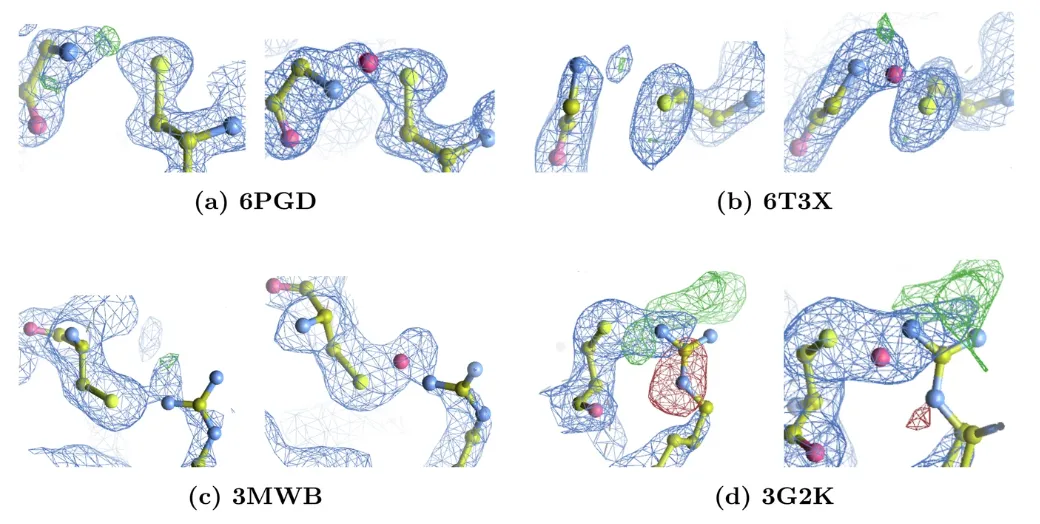
Durch explizite Modellierung und Neuverfeinerung konnte das ForschungsteamNach der Einführung von NOS-Bindungen verbesserte sich der Rwork/Rfree-Wert um durchschnittlich 0,5% und der unerklärte Elektronendichtepeak wurde deutlich reduziert.Für 3G2K gibt es in der ursprünglichen Struktur einen negativen Elektronendichtepeak um die Argininseitenkette, der nach der Umverteilung der Argininkonformation deutlich reduziert ist. Darüber hinaus gibt es in beiden Modellen positive Differenzpeaks in der Nähe der Argininseitenkette. Aufgrund ihrer großen Amplitude und der Anwesenheit von DMSO könnten sie Lösungsmittelmoleküle darstellen, die im aktuellen Modell nicht modelliert sind.
Strukturelle und thermodynamische Verifizierung
Um die Verbindung zwischen Arg-NOS-Cys und Gly-NOS-Cys weiter zu bestätigen, kombinierte das Forschungsteam die quantenmechanische Geometrieoptimierung mit der thermodynamischen Auswertung von vier repräsentativen Proteinkomplexen (6PGD, 6T3X, 3MWB und 3G2K), um die mögliche chemische Variabilität in vivo systematisch zu erklären.
In Bezug auf die Strukturüberprüfung liegt der SN-Abstand im optimierten NOS-Bindungsmodell zwischen 2,61 und 2,70 Å, was sehr nahe am Intervall von 2,63 bis 2,89 Å der ursprünglichen PDB-REDO-Struktur liegt.Simulationen ohne das überbrückende Sauerstoffatom führten zu einer signifikanten Erhöhung des SN-Abstands auf 3,36–4,26 Å, was darauf hindeutet, dass die experimentell beobachteten kürzeren SN-Abstände mit der Anwesenheit eines intermediären Sauerstoffatoms übereinstimmen.
Im Hinblick auf die thermodynamische Auswertung berechnete das Forschungsteam die Gibbs-Freienergie (ΔG) unter verschiedenen Protonierungszuständen und zeigte, dass alle NOS-Bindungsbildungsprozesse negativ sind.Dies deutet darauf hin, dass der Ersatz eines Wasserstoffatoms durch ein Sauerstoffatom zur Bildung der NOS-Bindung im simulierten Zustand thermodynamisch möglich ist. Die Größe von ΔG unterscheidet sich jedoch signifikant mit dem Protonierungszustand und zwischen den Arginin- und Glycin-basierten Komplexen. In beiden Systemen wird neutrales Glycin bzw. Arginin gegenüber den positiv geladenen Zuständen bevorzugt. Die Glycin-basierten Komplexe weisen leicht höhere ΔG-Werte auf. Obwohl diese Werte immer noch eine thermodynamisch günstige Assoziation implizieren, sind sie systematisch weniger exergonisch als die entsprechenden Argininkomplexe.
Zusammengenommen liefern diese strukturellen Ergebnisse übereinstimmende Beweise dafür, dassEs wurde gezeigt, dass die Arg-NOS-Cys- und Gly-NOS-Cys-Bindungen eher vernünftige kovalente Bindungen als einfache nichtgebundene Kontakte waren.Gleichzeitig deuten die Übereinstimmung zwischen den quantenmechanisch optimierten Geometrien und den kristallographischen Daten des Kristallsystems sowie die negativen freien Bildungsenergien stark darauf hin, dass diese Verbindungen in der relevanten Proteinumgebung strukturell und energetisch möglich sind.
Maschinelles Lernen eröffnet ein neues Kapitel in der mikroskopischen Welt der Proteine
Wie in der Studie erwähnt, hat die rasante Entwicklung von Technologien des maschinellen Lernens und der künstlichen Intelligenz bei der Lösung komplexer biochemischer Probleme eine Überlegenheit gegenüber herkömmlichen biochemischen Methoden bewiesen. Die geringen Rechenkosten und die hohe Effizienz haben die wissenschaftliche Forschungsgemeinschaft zu einer umfassenden Revolution der Produktionsmethoden veranlasst und maschinenlernbasierte Technologien gefördert, um ein größeres Potenzial bei der Entschlüsselung von Proteinfunktionen und der Identifizierung neuer Proteininteraktionen auszuschöpfen.
Zufällig veröffentlichten Kevin K. Yang et al. vom California Institute of Technology einen Artikel mit dem Titel „Machine learning-guided directed evolution for protein engineering“ in Nat. Methods.Durch den Vergleich zwischen gerichteter Evolution und maschinell lernengestützter gerichteter Evolution wird die Überlegenheit des maschinellen Lernens erklärt.Gleichzeitig listet der Artikel auch praktische Fälle auf, wie etwa die Optimierung der katalytischen Effizienz von Enzymen und der thermischen Stabilität von Cytochrom P450, und erwähnt verschiedene Methoden des maschinellen Lernens, wie etwa lineare Regression, Gauß-Prozess und Bayes-Optimierung.Es zeigt, dass maschinelles Lernen eine „datengesteuerte intelligente Navigation“ für das Protein-Engineering ermöglichen kann.Durch die Modellierung von Sequenz-Funktions-Beziehungen können die Effizienz und Erfolgsrate der gerichteten Evolution deutlich verbessert werden.
Papieradresse:
https://arxiv.org/pdf/1811.10775
Darüber hinaus wurde in einem von Rita Casadio et al. von der Universität Bologna in Italien veröffentlichten Artikel mit dem Titel „Maschinelle Lernlösungen zur Vorhersage von Protein-Protein-Interaktionen“ die Erforschung des maschinellen Lernens in der Proteinforschung ausführlich beschrieben.Es führt in die Anwendung maschineller Lernmethoden ein, einschließlich unüberwachtem und überwachtem Lernen bei Protein-Protein-Molekülinteraktionen (PPI).Wichtige Probleme hinsichtlich Datenqualität, Darstellung, Trainingsalgorithmen und Validierungsverfahren werden hervorgehoben.
Papieradresse:
https://wires.onlinelibrary.wiley.com/doi/full/10.1002/wcms.1618
Generell sind in der mikroskopischen Welt der Proteine noch viele Codes im Zusammenhang mit dem Leben verborgen. Die systematische, datengesteuerte Methode mit maschinellem Lernen als wichtigstem Mittel ist zweifellos wie ein Schlüssel, der die Tür zur mikroskopischen Welt der Proteine öffnet und die wissenschaftliche Forschungsgemeinschaft dazu inspiriert, die Funktion und Stabilität von Proteinen noch eingehender zu erforschen und zu untersuchen, wodurch die Grenzen der menschlichen Wahrnehmung des Lebens immer weiter durchbrochen werden.








