Command Palette
Search for a command to run...
Die Für ICML 2025 Ausgewählte Tsinghua-Universität/Renmin-Universität Schlug UniSim Vor, Einen Einheitlichen Biomolekulardynamik-Simulator

Die Gruppe von Professor Liu Yang von der Tsinghua-Universität und die Gruppe von Professor Huang Wenbing von der Gaoling School of Artificial Intelligence an der Renmin-Universität China haben gemeinsam einen einheitlichen biomolekularen zeitvergröberten Dynamiksimulator UniSim vorgeschlagen.Diese Methode erhält ein einheitliches Allatom-Darstellungsmodell durch hybrides Vortraining aus Rauschunterdrückung und Kraftfeld anhand einer großen Menge 3D-Molekülstrukturdaten, lernt das Transfervektorfeld von Molekülen in einem langen Zeitschritt basierend auf einem stochastischen interpolierenden generativen Rahmen und führt einen kraftgesteuerten Kern ein, um sich schnell an unterschiedliche chemische Umgebungen anzupassen. UniSim ist das erste System, das ein einheitliches, zeitvergröbertes Dynamiksimulationsframework für Molekültypen (kleine Moleküle, Peptide, Proteine) und chemische Umgebungen realisiert.Es hat die praktische Anwendung von Deep Learning im Bereich der molekularen Simulation gefördert.
Die entsprechenden Ergebnisse wurden für ICML 2025 unter dem Titel „UniSim: A Unified Simulator for Time-Coarsened Dynamics of Biomolecules“ ausgewählt.

Papieradresse:
Weitere Artikel zu den Grenzen der KI:
https://go.hyper.ai/UuE1o
Warum brauchen wir einen einheitlichen zeitlichen Vergröberungssimulator?
Die Forscher sind der Ansicht, dass es im Bereich der Molekulardynamiksimulation sinnvoll und notwendig ist, einen einheitlichen Zeitvergröberungssimulator zu bauen.Einerseits ist ein einheitlicher Modellierungsrahmen die Grundlage für die kollaborative Simulation molekularer Systeme.Bei der Simulation komplexer Systeme wie Protein-Ligand-Wechselwirkungen koexistieren Proteine und kleine Moleküle häufig in derselben physikalischen Umgebung. Ist das Modell nur auf einen bestimmten Molekültyp anwendbar, ist es schwierig, das Kopplungsverhalten zwischen beiden auf atomarer Ebene präzise nachzubilden. Daher können Simulatoren mit einheitlichen Darstellungsmöglichkeiten Moleküle verschiedener Typen gleichzeitig im selben Modellrahmen verarbeiten und so eine solide Grundlage für die Modellierung multimolekularer Komplexe bieten.
Andererseits hilft ein einheitliches Modell dabei, die strukturellen und dynamischen Daten verschiedener Molekültypen zu integrieren und so die Generalisierungs- und Übertragungsmöglichkeiten des Modells zu verbessern.Derzeit verfügbare Daten zu molekularen Trajektorien sind äußerst selten und ungleichmäßig verteilt. Verschiedene Datentypen wie Proteine, Peptide und kleine Moleküle haben ihre eigenen Stärken. Wenn sie alle am Vortraining und Lernen im selben Modell teilnehmen können, wird das Gesamtverständnis des Modells für Strukturen auf atomarer Ebene deutlich verbessert und es verfügt über stärkere Fähigkeiten zur domänenübergreifenden Migration.
gleichzeitig,Die Einführung einer Zeitvergröberungssimulation ist ebenfalls eine wichtige Möglichkeit zur Verbesserung der Simulationseffizienz.Herkömmliche Molekulardynamiksimulationen basieren auf extrem kleinen Zeitschritten (z. B. Femtosekunden), um schrittweise fortzuschreiten. Dies ist rechenintensiv und erschwert die Abbildung langfristiger Verhaltensweisen wie der Proteinfaltung. Die Zeitvergröberungsmethode lernt die Abbildungsbeziehung vom aktuellen zum zukünftigen Zustand direkt. Unter der Voraussetzung der Wahrung der physikalischen Konsistenz kann sie schnell Trajektorien in einem Zeitmaßstab generieren, der deutlich größer ist als die herkömmliche Schrittweite. Dies verbessert die Simulationseffizienz erheblich und ermöglicht Langzeitsimulationen in praktikabler Zeit.
Einheitliche Darstellung: Lösung des Problems der Charakterisierung von Molekülen mit mehreren Skalen und Typen
Obwohl einheitliche All-Atom-Darstellungsmodelle den Grundstein für Dynamiksimulationen über Molekülarten hinweg bilden,Die Implementierung eines solchen Modells ist jedoch noch mit folgenden technischen Herausforderungen verbunden:
* Erstens reichen molekulare Systeme von kleinen organischen Molekülen mit Dutzenden von Atomen bis hin zu Proteinmakromolekülen mit Tausenden von Atomen, wobei es große Unterschiede in der Größe und komplexe und vielfältige Strukturen gibt.Wenn alle Atome direkt zum Training verwendet werden, verfügt das Modell über unterschiedliche Aufmerksamkeitsmechanismen für unterschiedliche Molekültypen, wodurch die Fähigkeit des Modells zur gegenseitigen Übertragung beeinträchtigt wird.
* Zweitens ist die Grundlage für die Erreichung einer einheitlichen Darstellung aller Atome die Verwendung eines einheitlichen Vokabulars auf atomarer Ebene.Ein intuitiver Ansatz besteht darin, das Periodensystem direkt als Vokabular für die Einbettungsdarstellung zu verwenden. Dieser Ansatz ignoriert jedoch die regulären Einheiten, die in Peptiden und Proteinen in großer Zahl vorkommen, wie z. B. Substrukturen wie natürliche Aminosäuren, was zu einer schlechten Leistung bei Proteintypdaten führt.
* Um die Darstellung von Molekülen in verschiedenen Zuständen vollständig zu erlernen, werden schließlich große Mengen stationärer und nicht-stationärer 3D-Molekülstrukturdaten in den vorab trainierten Datensatz aufgenommen.Das gängige Paradigma für das Vortraining instabiler Moleküle besteht darin, die auf Atome wirkenden Kräfte zu erlernen. Unterschiedliche Datensätze verwenden jedoch unterschiedliche Kraftfeldparameter bei der Berechnung atomarer Kraftfelder, und es kommt zu einer Fehlausrichtung der Beschriftungsdaten.
Um eine einheitliche Modellierung zu erreichen, führt UniSim drei Schlüsseltechnologien ein, um die oben genannten Probleme zu lösen:
* Gradienten-Umgebungs-Subgraph: vernünftiger Ausgleich der molekularen Skala
In der Datenvorverarbeitung werden die 3D-Strukturdaten großer Moleküle (mit mehr als 1.000 Atomen) segmentiert.Min < rmax Während der Vorverarbeitung wird ein beliebiges Atom im Molekül zufällig ausgewählt und als Mittelpunkt der Kugel genommen.Min und rmax Erstellen Sie eine Kugel mit RadiusDie in der kleinen Kugel enthaltenen Atome werden als Gradienten-Teilgraph betrachtet, und die in der großen Kugel enthaltenen Atome werden als Umgebungs-Teilgraph betrachtet.Basierend auf der physikalischen Annahme, dass die interatomare Kraft im Allgemeinen exponentiell mit der Entfernung abnimmt, wenn rmax- RMin Bei richtiger Auswahl ist die Wechselwirkung zwischen Atomen außerhalb des Umgebungs-Subgraphen im ursprünglichen Molekül und Atomen im Gradienten-Subgraphen vernachlässigbar. Daher wird während des Trainings der Umgebungs-Subgraph anstelle des ursprünglichen Moleküls als Eingabe verwendet, und nur der Gradienten-Subgraph wird in die Berechnung der Verlustfunktion einbezogen. Dadurch wird der Umfang der Molekülstrukturdaten angemessen ausgeglichen und die Kreuztransferfähigkeit des Modells verbessert.
* Atom-Embedding-Erweiterung: Erhalten Sie eine verfeinerte atomare Darstellung
Diese Studie basiert auf dem Periodensystem der Elemente.Einführung mehrerer lernbarer diskreter Einbettungsdarstellungen für dasselbe Element als erweitertes Vokabular,Es wird verwendet, um die reguläre Substruktur zu erfassen, in der sich Atome befinden. Basierend auf einem einfachen Graph-Neuralnetzwerk integriert UniSim die Nachbarschaftsinformationen jedes Atoms, ermittelt die Wahrscheinlichkeit jeder eingebetteten Darstellung im erweiterten Vokabular, das dem Atom entspricht, und erhält die erweiterte eingebettete Darstellung des Atoms durch gewichtete Summation.Diese Darstellung gleicht die Genauigkeit auf atomarer Ebene mit regelmäßigen Unterstrukturen innerhalb bestimmter Molekülarten aus, was zu einer effizienten und detaillierten atomaren Darstellung führt.
* Multi-Head-Hybrid-Pre-Training: Hybrides Lernen von Daten mit unterschiedlichen Molekülzuständen und Label-Verteilungen
UniSim verwendet die folgende Methode, um stationäre und nichtstationäre Molekülstrukturen gemeinsam zu erlernen: Für stationäre Daten verwendet der Artikel das Denoising-Pretraining-Paradigma, um die verrauschten Daten zu entrauschen und die atomare Darstellung zu erlernen; für nichtstationäre Daten lernt das Modell direkt das konservative Kraftfeld, und unterschiedliche Kraftfeldparameter entsprechen unterschiedlichen Ausgabeköpfen, wodurch die durch unterschiedliche Beschriftungsverteilungen verursachten Fehler vermieden werden.
Der Artikel verwendet TorchMD-NET als grundlegendes Graph-Neural-Network-Modell, das die SO(3)-Äquivarianz erfüllt. Basierend auf den oben genannten wichtigen Vortrainingstechniken,Das Vortraining mit umfangreichen 3D-Moleküldaten aus mehreren Quellen wurde abgeschlossen und die effektive Konstruktion eines einheitlichen atomaren Darstellungsmodells wurde erreicht.
Vektorfeldmodelle: Lernen langfristiger Zustandsübergänge aus Trajektorien
Herkömmliche molekulardynamische Simulationen sind durch Integrationsschritte von wenigen Femtosekunden begrenzt, was die effiziente Untersuchung von Langzeitverhalten wie der Proteinfaltung erschwert. UniSim verwendet ein stochastisches Interpolationsframework und verbindet ein geometrisches Vektorperzeptron als Vektorfeldmodell nach einem vortrainierten All-Atom-Darstellungsmodell.Das Modell erreicht eine durchgängige, zeitvergröberte Dynamikmodellierung, indem es das Transfervektorfeld zwischen Molekülzuständen in langen Zeitschritten lernt.
Während des Trainings werden Paare molekularer Konformationen, die durch einen bestimmten Zeitschritt in der realen dynamischen Trajektorie getrennt sind, als Trainingsbeispiele ausgewählt, zufällige Störungen in den Interpolationspfad eingeführt und das Geschwindigkeitsfeld (Velocity) und der Rauschunterdrücker (Denoiser) gemeinsam erlernt, um eine Trajektoriengenerierung in kontinuierlicher Zeit zu erreichen. Im Vergleich zur herkömmlichen numerischen Integration kann UniSim die Simulationseffizienz deutlich verbessern und den Engpass der herkömmlichen Simulation im Zeitmaßstab überwinden.
Kraftgesteuerte Kerne: Schnelle Anpassung an komplexe chemische Umgebungen
Die Molekulardynamik weist unter verschiedenen Lösungsmittel-, Temperatur- und Druckbedingungen unterschiedliche potenzielle Energieoberflächen auf, die die Verteilung der erzeugten Konformationen stark beeinflussen.Zu diesem Zweck führt UniSim einen Kraftführungskernel ein, um ein virtuelles Zwischenkraftfeld im Rahmen der Zufallsdifferenz zu definieren und so die Trajektorienabtastung zu steuern.Dieses Zwischenkraftfeld entspricht dem realen MD-Kraftfeld an beiden Enden des Erzeugungspfads (d. h. dem Anfangszustand und dem Endzustand) und ist so konzipiert, dass es in hohem Maße mit den physikalischen Vorbedingungen übereinstimmt, sodass die erzeugte Konformation besser mit der Boltzmann-Verteilung unter dem Zielkraftfeld übereinstimmt.
Durch die Anpassung des Zwischenkraftfelds muss UniSim die Parameter des vortrainierten Modells und des Vektorfeldmodells nicht ändern.Es muss lediglich ein steckbarer Kraftführungskernel erlernt werden, damit sich das Zielkraftfeld effizient an neue chemische Umgebungen anpassen kann.Verbessert effektiv die Generalisierungs- und Migrationsfunktionen des Modells.
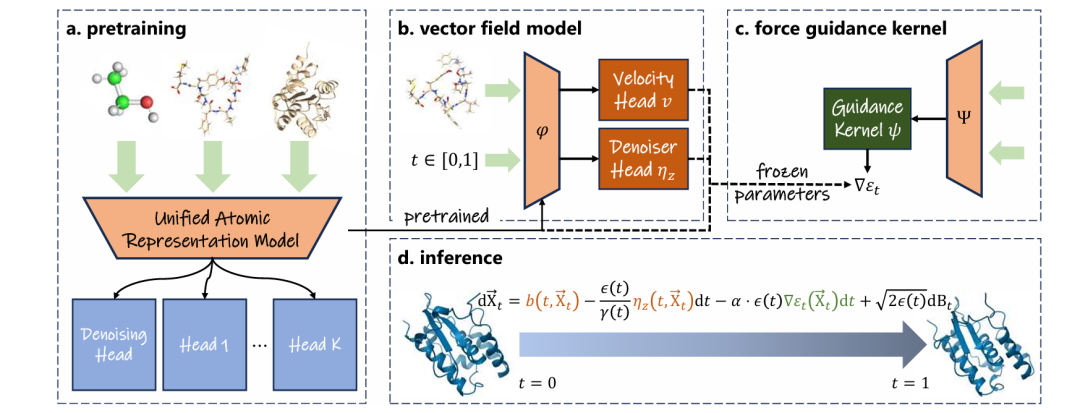
Experimentelle Überprüfung: mehrere Molekültypen
Um die Vielseitigkeit von UniSim an verschiedenen Molekültypen zu überprüfen,Die Forscher werteten in einer Vorwärtssimulationsaufgabe systematisch Daten mehrerer Molekültypen aus, darunter drei Molekültypen: kleine Moleküle, Peptide und Proteine.Durch einen Vergleich mit dem Deep-Learning-Modell in diesem Bereich, das ebenfalls eine zeitvergröberte Dynamiksimulation durchführt, soll im Experiment untersucht werden, ob die vereinheitlichte atomare Darstellung dazu beitragen kann, das Verständnis des Modells für molekulare Zustände und modalübergreifende Generalisierungsfähigkeiten zu verbessern, und wie sich die Einbeziehung kraftgesteuerter Kerne auf die Leistung des Modells in Bezug auf Schlüsselindikatoren wie die Rationalität der unter dem Zielkraftfeld erzeugten Konformationen und die Verteilungsähnlichkeit auswirkt.
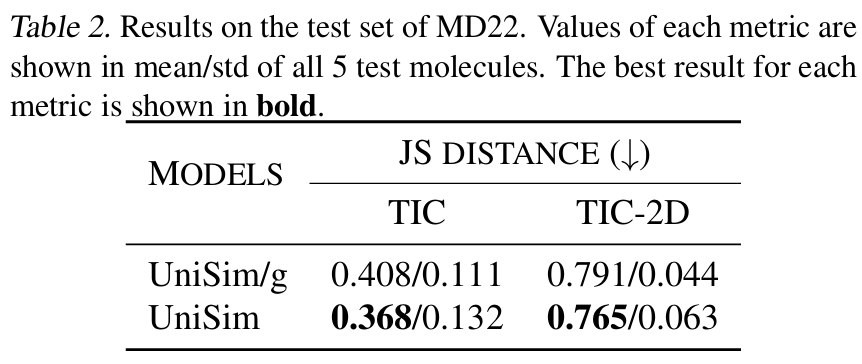
Die Ergebnisse zeigen, dass UniSim bei allen Molekültypen eine umfassende Überlegenheit erreicht hat.Es weist eine gute Leistung bei der Verteilungsähnlichkeit auf und weist eine deutliche Verbesserung beim wichtigsten Indikator für die Konformationsrationalität (Val-CA) auf. Es ist zu beachten, dass bei der Aufgabe der Vorwärtssimulationsgenerierung jede Konformation in der Trajektorie durch Autoregression generiert wird, die einen enormen kumulativen Fehler aufweist, sodass es ziemlich schwierig ist, die Rationalität der Konformation zu verbessern.
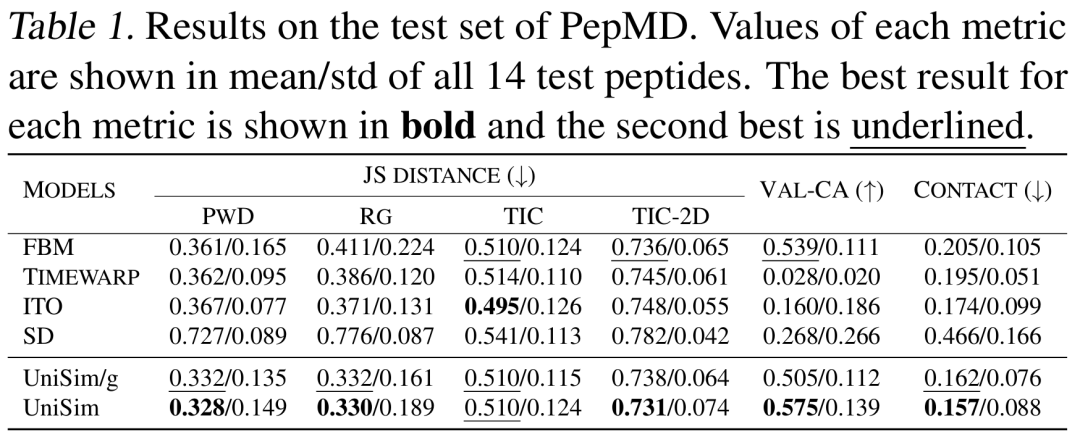
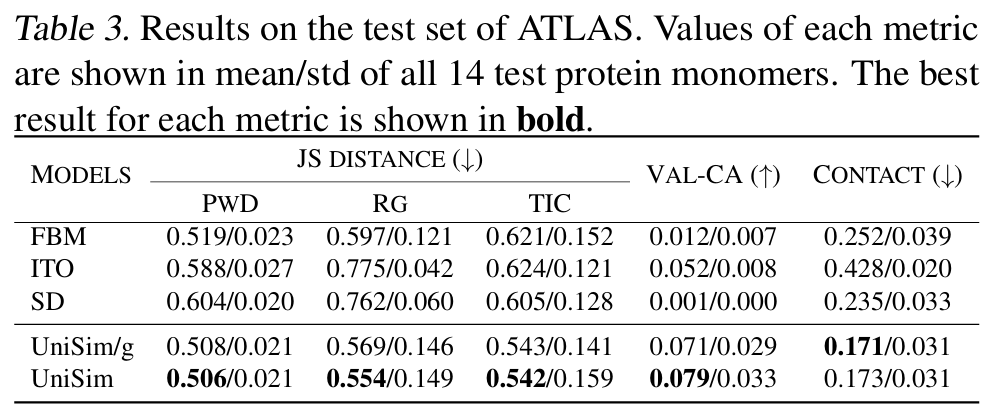
Bei der Vorwärtssimulation von Peptiden und ProteinenIm Vergleich zu bestehenden Methoden wie FBM, ITO und SD ist UniSim bei Indikatoren wie Verteilungsähnlichkeit (TIC-2D), Strukturrationalität (VAL-CA) und Kontaktkartenfehler (CONTACT) führend. Insbesondere nach der Einführung des kraftgeführten Kerns behält UniSim das ursprüngliche Niveau bei Indikatoren wie Verteilungsähnlichkeit bei, hat sich jedoch bei wichtigen Indikatoren der Konformationsrationalität deutlich verbessert. Gleichzeitig kann UniSim in komplexen Proteinsystemen Energiebarrieren überwinden und mehrere metastabile Zustände in nur Hunderten von Vorwärtssimulationsschritten abdecken, was neue Wege für die effiziente Simulation großer Biomoleküle eröffnet.
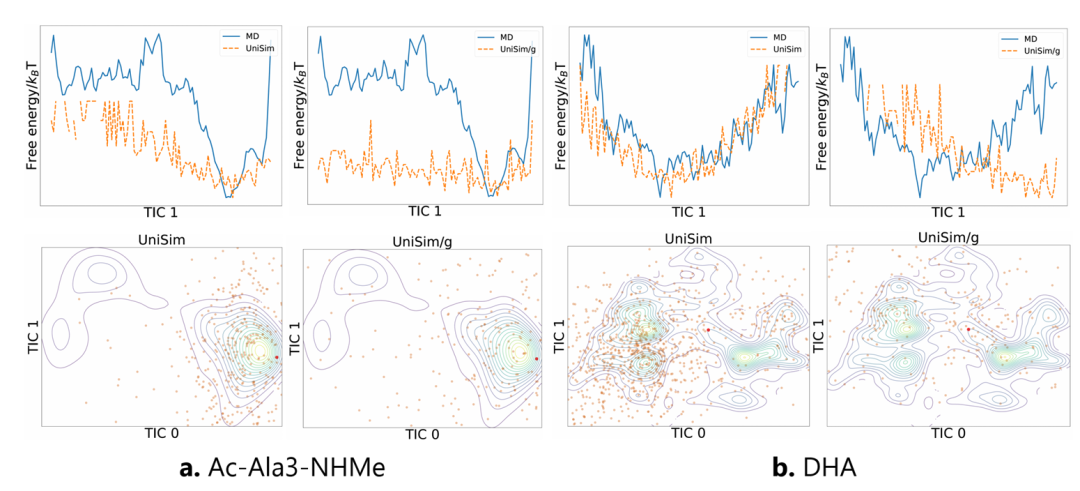
Alanin-Dipeptid-Fallstudie
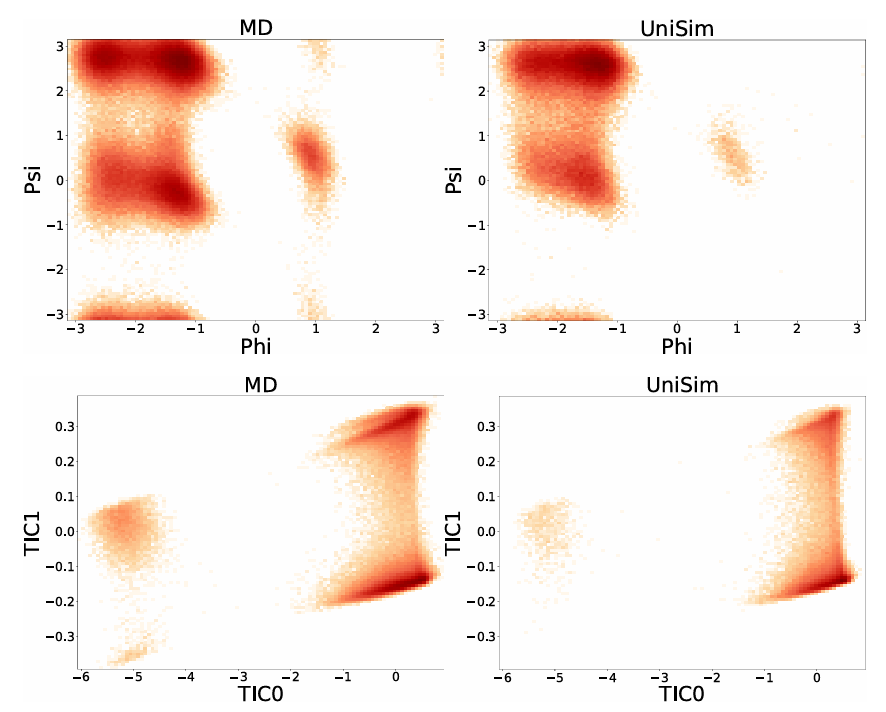
Um außerdem die Stabilität von UniSim in langfristigen molekulardynamischen Simulationen zu untersuchen, haben die Forscher das Modell anhand des klassischen Systems Alanin-Dipeptid fein abgestimmt und Langzeitsimulationen mit 100.000 Schritten durchgeführt.Durch Vergleich mit MD-Ergebnissen konnte UniSim erfolgreich fünf bekannte metastabile Schlüsselzustände reproduzieren.Die freie Energielandschaft des Alanin-Dipeptids im dynamischen Prozess wurde genau wiederhergestellt, wodurch die Stabilität und physikalische Konsistenz des Modells bei der Langzeitsimulation vollständig bestätigt wurde.
Ausblick
UniSim ist das erste Framework, das eine einheitliche zeitlich grobkörnige Dynamiksimulation über Molekültypen und chemische Umgebungen hinweg ermöglicht.Dies eröffnet einen praktikablen Weg für die breite Anwendung von Deep Learning in der Arzneimittelforschung, im Proteindesign und anderen Szenarien. Die Forscher wiesen außerdem darauf hin, dass folgende Bereiche künftig weiter erforscht werden könnten:
* Ein effizienterer Mechanismus zur kreuzmodalen Konformationsoptimierung zur Verbesserung der Effektivität der generierten Proben;
* Trajektorienmodellierung auf längeren Zeitskalen, um komplexe biophysikalische Mechanismen aufzudecken;
* Erforschen Sie die dynamischen Mechanismen in komplexen Systemen und konzentrieren Sie sich dabei auf intermolekulare Wechselwirkungen.








