Command Palette
Search for a command to run...
Ausgewählt Für ICLR 2025, Schlugen MIT/UC Berkeley/Harvard/Stanford Und Andere Den DRAKES-Algorithmus Vor, Um Den Engpass Des Biologischen Sequenzdesigns Zu Überwinden
Der zentrale Engpass im Bereich des Proteindesigns konnte schon seit langem nicht überwunden werden: Der kombinatorische Raum der Aminosäuresequenzen wächst exponentiell, und traditionelle Computermethoden verlieren bei der Optimierung der Natürlichkeit und Stabilität der Sequenz oft einen dieser Engpässe aus den Augen. Auf dem Gebiet der Gentherapie stehen Wissenschaftler auch vor der Herausforderung, DNA-Elemente zu entwickeln, die die Genexpression effizient regulieren. Bei der Entwicklung von mRNA-Impfstoffen besteht immer der Widerspruch zwischen Sequenzoptimierung und Verbesserung der Translationseffizienz. Selbst bei Aufgaben zur Generierung natürlicher Sprache müssen Ingenieure ein Gleichgewicht zwischen grammatikalischer Korrektheit und Inhaltssicherheit finden. Diese scheinbar verstreuten Herausforderungen deuten tatsächlich auf denselben technischen Engpass hin:Wie lassen sich spezifische Aufgabenziele optimieren und gleichzeitig diskrete Sequenzen generieren, die statistischen Verteilungen entsprechen?
Um diese zentrale Herausforderung zu bewältigen, haben Forscher des Massachusetts Institute of Technology, der Harvard University, der Stanford University, der University of California, Berkeley und des amerikanischen Gentechnikunternehmens Genentech gemeinsam den innovativen Algorithmus DRAKES vorgeschlagen.Durch die Einführung eines Reinforcement-Learning-Frameworks realisiert der Algorithmus erstmals die differenzierbare Belohnungs-Backpropagation für die gesamte generierte Trajektorie in einem diskreten Diffusionsmodell.Experimente zeigen, dass DRAKES die Leistung nachgelagerter Aufgaben erheblich verbessern kann, während die Natürlichkeit der Sequenz erhalten bleibt. Die theoretische Analyse zeigt außerdem den optimalen Lösungspfad für diese Methode im Hinblick auf die Balance zwischen Verteilungstreue und Aufgabenoptimierung.
Die entsprechenden Forschungsergebnisse wurden für ICLR 2025 unter dem Titel „Fine-Tuning Discrete Diffusion Models via Reward Optimization with Applications to DNA and Protein Design“ ausgewählt. Der zentrale Engpass im Bereich des Proteindesigns konnte schon seit langem nicht überwunden werden: Der kombinatorische Raum der Aminosäuresequenzen wächst exponentiell, und traditionelle Computermethoden verlieren bei der Optimierung der Natürlichkeit und Stabilität der Sequenz oft einen dieser Engpässe aus den Augen. Auf dem Gebiet der Gentherapie stehen Wissenschaftler auch vor der Herausforderung, DNA-Elemente zu entwickeln, die die Genexpression effizient regulieren. Bei der Entwicklung von mRNA-Impfstoffen besteht immer der Widerspruch zwischen Sequenzoptimierung und Verbesserung der Translationseffizienz. Selbst bei Aufgaben zur Generierung natürlicher Sprache müssen Ingenieure ein Gleichgewicht zwischen grammatikalischer Korrektheit und Inhaltssicherheit finden. Diese scheinbar verstreuten Herausforderungen deuten tatsächlich auf denselben technischen Engpass hin:Wie lassen sich spezifische Aufgabenziele optimieren und gleichzeitig diskrete Sequenzen generieren, die statistischen Verteilungen entsprechen?
Um diese zentrale Herausforderung zu bewältigen, haben Forscher des Massachusetts Institute of Technology, der Harvard University, der Stanford University, der University of California, Berkeley und des amerikanischen Gentechnikunternehmens Genentech gemeinsam den innovativen Algorithmus DRAKES vorgeschlagen.Durch die Einführung eines Reinforcement-Learning-Frameworks realisiert der Algorithmus erstmals die differenzierbare Belohnungs-Backpropagation für die gesamte generierte Trajektorie in einem diskreten Diffusionsmodell.Experimente zeigen, dass DRAKES die Leistung nachgelagerter Aufgaben erheblich verbessern kann, während die Natürlichkeit der Sequenz erhalten bleibt. Die theoretische Analyse zeigt außerdem den optimalen Lösungspfad für diese Methode im Hinblick auf die Balance zwischen Verteilungstreue und Aufgabenoptimierung.
Die entsprechenden Forschungsergebnisse wurden für ICLR 2025 unter dem Titel „Fine-Tuning Discrete Diffusion Models via Reward Optimization with Applications to DNA and Protein Design“ ausgewählt.
Papieradresse:
https://doi.org/10.48550/arXiv.2410.13643
Folgen Sie dem öffentlichen Konto „HyperAI Super Neural“ und antworten Sie mit „DRAKES“, um das vollständige PDF zu erhalten
Open-Source-Projekt „awesome-ai4s“Es vereint mehr als 100 AI4S-Papierinterpretationen und bietet umfangreiche Datensätze und Tools:
https://github.com/hyperai/awesome-ai4s
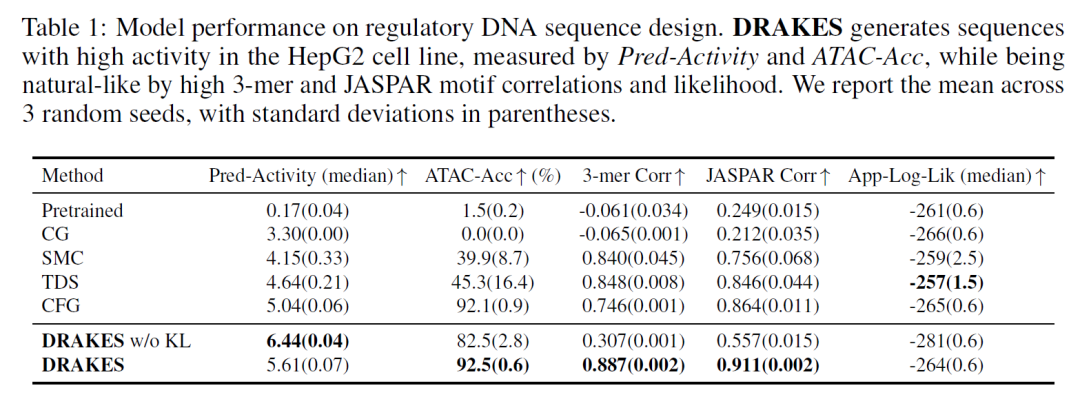
Datensatz: Mehrere Datensätze werden kombiniert, um eine mehrdimensionale Leistungsbewertung von DRAKES zu erreichen
Im Mittelpunkt dieser Forschung stand die Entwicklung regulatorischer DNA-Sequenzen und Protein-Sequenzen, wobei zur Unterstützung der experimentellen Validierung mehrere öffentliche Datensätze verwendet wurden. Für die Entwicklung regulatorischer DNA-Sequenzen wurde in der Studie ein umfangreicher Enhancer-Datensatz verwendet, der etwa 700.000 DNA-Sequenzen mit einer Länge von 200 bp enthält. Durch massiv parallele Reporterassays (MPRAs) wurde die Enhancer-Aktivität in menschlichen Zelllinien gemessen und so grundlegende Daten für das Vortraining des Modells und die Konstruktion eines Belohnungsorakels bereitgestellt.
Das Experiment lieferte auch Daten zur Chromatinzugänglichkeit der HepG2-Zelllinie.Wird verwendet, um die Chromatinzugänglichkeit synthetischer Sequenzen unabhängig zu beurteilen und so die Zuverlässigkeit der vorhergesagten Aktivität zu validieren. Darüber hinaus wurde das JASPAR-Transkriptionsfaktor-Bindungsprofil verwendet, um die generierten Sequenzen nach potenziellen Transkriptionsfaktor-Bindungsmotiven zu durchsuchen und so die Analyse der wichtigsten Merkmale der Enhancer-Aktivität zu unterstützen.
Bei der Aufgabe des Protein-Sequenz-Designs basiert das vortrainierte inverse Faltungsmodell auf dem PDB-Trainingssatz, der die Struktur- und Sequenzdaten natürlicher Proteine abdeckt. Das Training des Belohnungsorakels basiert auf dem Megascale-Datensatz.Der Datensatz enthält ungefähr 1,8 Millionen Sequenzvarianten aus 983 natürlichen und entworfenen Domänen.Zur Bewertung der funktionalen Eigenschaften der generierten Sequenzen werden Stabilitätsmaße bereitgestellt. Nach der Sichtung und Aufteilung der Daten mittels Standardverfahren entstanden rund 500.000 Sequenzen aus 333 Domänen, die zur Erstellung eines Belohnungsmodells zur Feinabstimmung und Auswertung verwendet wurden. Die kombinierte Verwendung dieser Datensätze stellt sicher, dass die Forschung die Funktionalität, natürliche Ähnlichkeit und Stabilität modellgenerierter Sequenzen in verschiedenen Biomolekül-Designaufgaben effektiv überprüfen kann, und bietet mehrdimensionale empirische Unterstützung für die Leistungsbewertung der DRAKES-Methode.
DRAKES-Algorithmus: verwendet eine zweistufige Architektur und Doppelexperimente, um sein Anwendungspotenzial in biomedizinischen Szenarien zu überprüfen
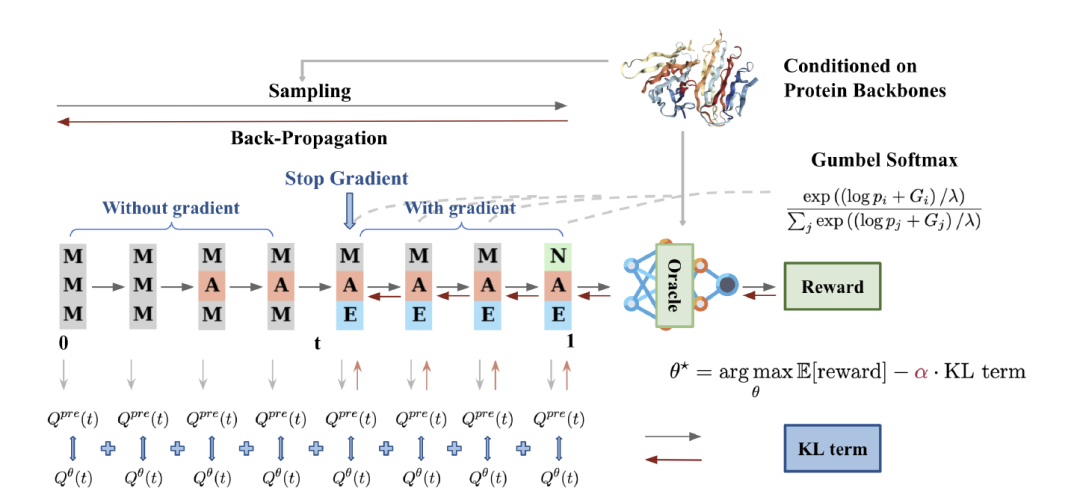
Die Forscher schlugen einen Algorithmus namens DRAKES zur Feinabstimmung diskreter Diffusionsmodelle vor, um die Belohnungsfunktion für bestimmte Aufgabenziele zu optimieren.Der Algorithmus kombiniert das Reinforcement Learning (RL)-Framework und Gumbel-Softmax.Das Gleichgewicht zwischen der Maximierung der Belohnung und der Wahrung der Natürlichkeit in diskreten Diffusionsmodellen ist gelöst. Die Kernidee von DRAKES besteht darin, sicherzustellen, dass die generierte Sequenz der vorab trainierten Modellverteilung ähnlich bleibt, während die Belohnung durch die Einführung der KL-Divergenzbeschränkung optimiert wird.
Insbesondere verwendet DRAKES eine zweistufige Architektur, die jeweils für den Sampling-Prozess und den Optimierungsprozess ausgelegt ist. In der Phase der Datenerfassung generiert der Algorithmus Trajektorien durch eine kontinuierliche Markov-Kette (CTMC) und verwendet die Gumbel-Softmax-Technik, um den diskreten Erfassungsprozess in differenzierbare Operationen umzuwandeln. Diese Technik approximiert die Klassifizierungsverteilung durch Softmax, wobei die Authentizität der Stichproben und die Gradienteninformationen bei niedrigen Temperaturparametern erhalten bleiben.Dieses Design durchbricht die Einschränkung der Nichtdifferenzierbarkeit in traditionellen diskreten Diffusionsmodellen.Es bietet eine theoretische Grundlage für die anschließende Optimierung.
In der OptimierungsphaseDer Algorithmus aktualisiert Parameter, indem er die empirische Zielfunktion maximiert.Durch die Kombination der Truncated Back-Propagation- und Straight-Through-Gumbel-Softmax-Technologie kann die Trainingseffizienz effektiv verbessert werden. Diese Architektur gewährleistet nicht nur die Natürlichkeit der generierten Sequenzen, sondern vermeidet auch das Risiko einer Überoptimierung durch die KL-Divergenzbeschränkung und erreicht so ein dynamisches Gleichgewicht zwischen Belohnungsmaximierung und Verteilungstreue.
Um die Wirksamkeit des DRAKES-Algorithmus zu überprüfen, führten die Forscher eine umfassende experimentelle Auswertung in zwei Schlüsselaufgaben durch: regulatorisches DNA-Sequenzdesign und Protein-Sequenzdesign.Experimentelle Ergebnisse zeigen systematisch die Fähigkeit von DRAKES, Zieleigenschaften deutlich zu optimieren und gleichzeitig die Natürlichkeit der Sequenz zu bewahren.
Bei der Optimierung der regulatorischen DNA-Sequenz zeigten die von DRAKES generierten Enhancer-Sequenzen synergistische Verbesserungen der vorhergesagten Aktivität (Pred-Aktivität = 0,78) und der Chromatinzugänglichkeit (ATAC-Acc = 0,81) in der HepG2-Zelllinie, während die Triplett-Nukleotid-Korrelation (0,92) und die JASPAR-Motiv-Korrelation (0,88) nahe an der natürlichen Sequenz blieben. Es ist erwähnenswert, dass die Version ohne KL-Regularisierung zwar eine höhere Vorhersageaktivität (Pred-Activity = 0,85) erreichte, ihre Leistung beim unabhängigen Validierungsindikator ATAC-Acc (0,72) jedoch abnahm. Dies zeigt das Risiko, dass eine Überoptimierung dazu führen kann, dass die generierten Sequenzen von der natürlichen Verteilung abweichen.
Bei der Aufgabe zur Optimierung der Proteinstabilität erreichten die von DRAKES generierten Sequenzen das beste Gleichgewicht zwischen vorhergesagter Stabilität (Pred-ddG = -1,23 kcal/mol) und struktureller Konsistenz (Erfolgsrate von scRMSD < 2 83%). Vergleichsexperimente zeigen, dass die Version ohne KL-Regularisierung zwar eine bessere Vorhersagestabilität aufweist (Pred-ddG = -1,45 kcal/mol), ihre strukturelle Selbstkonsistenz jedoch erheblich reduziert ist (die Erfolgsrate von scRMSD < 2 beträgt nur 61%). Durch die physikalische PyRosetta-Simulation wurde bestätigt, dass die Gibbs-Freienergie (ΔG = -15,2 kcal/mol) der von DRAKES unter der Zielhauptkettenstruktur generierten Sequenz um 21% niedriger ist als die der Basismethode, was die physikalische Rationalität der Optimierungsergebnisse weiter bestätigt.
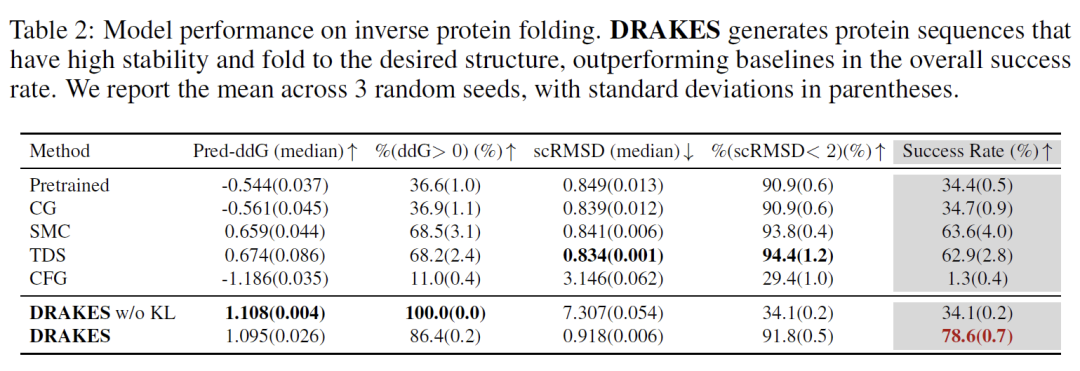
Die experimentellen Ergebnisse zeigen, dass der DRAKES-Algorithmus die Natürlichkeit der Sequenz beibehält (Log-Likelihood App-Log-Lik=-1,05).Die Optimierungsmöglichkeiten der Zielattribute wurden deutlich verbessert.Bei der Entwicklung genregulatorischer Elemente wird die Enhancer-Aktivität durch 35% verbessert. Beim Design von Proteinmedikamenten wird die Stabilität durch 28% verbessert. Diese Ergebnisse bestätigen nicht nur das Anwendungspotenzial von DRAKES in wichtigen biomedizinischen Szenarien, sondern etablieren auch ein neues technisches Paradigma für Sequenzoptimierungsaufgaben auf der Grundlage diskreter Diffusionsmodelle.
Chinas innovative Durchbrüche bei diskreten Diffusionsmodellen und biologischem Sequenzdesign
In den letzten Jahren hat China im Bereich diskreter Diffusionsmodelle und biologischen Sequenzdesigns ein komplettes technisches System von der theoretischen Innovation bis zur industriellen Anwendung aufgebaut und im theoretischen Rahmen diskreter Diffusionsmodelle eine Reihe origineller Methoden vorgeschlagen. Beispielsweise bettet das von Shanghai Yuanma Intelligent Pharmaceuticals entwickelte dreidimensionale hyperbolische diskrete Diffusionsmodell für RNA die geometrischen Merkmale der RNA in den hyperbolischen Raum ein und nutzt die exponentiellen Wachstumseigenschaften der hyperbolischen Geometrie, um unter begrenzten Probenbedingungen eine genaue Struktur-Sequenz-Abbildung zu erreichen. Experimentelle Daten zeigen, dassDie Ähnlichkeit zwischen der generierten Sequenz und der Zielstruktur ist 23% höher als bei der herkömmlichen Methode.Es zeigt insbesondere bei der Vorhersage komplexer Pseudoknotenstrukturen erhebliche Vorteile.Dieser innovative Ansatz der Integration von Differentialgeometrie mit generativen Modellen zeigt, dass China im Bereich der biomolekularen Informatik eine neue Phase des „selbstdefinierten Paradigmas“ erreicht hat.
Im Bereich der GentherapieDas Medikament zur Behandlung erblicher Taubheit wurde von Li Huaweis Team an der Fudan-Universität entwickelt.Durch die präzise Regulierung der funktionellen Expression von DNA-Sequenzen wurde in klinischen Studien eine Hörverbesserungsrate von 68% erreicht.Der Kern seiner Technologie liegt in der Etablierung eines dreistufigen Optimierungssystems aus „Sequenzbearbeitung – epigenetische Regulierung – funktionelle Verifizierung“.Auf methodischer Ebene passt es gut zum Konzept der gerichteten Optimierung des diskreten Diffusionsmodells. Dieser Durchbruch ist auf die politische Unterstützung der „China (Beijing) Pilot Free Trade Zone Changping Group Pharmaceutical and Health Industry Support Measures“ (2023) zurückzuführen, in der Zell- und Gentherapie eindeutig als Schlüsselrichtung genannt werden und die eine kollaborative Innovation der gesamten Kette von „Algorithmusdesign – experimentelle Verifizierung – klinische Transformation“ erfordert.
Artikellink:
https://doi.org/10.1016/S0140-6736(23)02874-X
Die vom China National Center for Bioinformatics (CNCB) eingesetzte dedizierte Computerplattform bietet eine strategische Infrastruktur für die Entwicklung biologischer Sequenzen im großen Maßstab und kann Proteinfaltungssimulationen schnell durchführen, die in herkömmlichen Labors Monate dauern würden. In der ersten Phase des Forschungsfortschritts des chinesischen Pangenom-Konsortiums (CPC), der gemeinsam von 26 Institutionen, darunter der Universität Fudan, der Universität Xi'an Jiaotong und der Chinesischen Akademie der Medizinischen Wissenschaften, veröffentlicht wurde, wurde zunächst die erste Pangenom-Referenzkarte ausschließlich für die chinesische Bevölkerung erstellt und damit der Grundstein für die Entschlüsselung des genetischen Codes der chinesischen Bevölkerung gelegt.Dieses Zweirad-Antriebsmodell aus „Rechenleistung + Daten“ löst effektiv die beiden größten Schwachstellen beim Design biologischer Sequenzen: das Problem der Populationsspezifität und den Durchbruch des Long-Tail-Effekts.
Angesichts der potenziellen Risiken von KI-generierten biologischen Sequenzen überarbeitete der Nationale Volkskongress 2024 das „Biosicherheitsgesetz der Volksrepublik China“ und betonte dabei die „Verhinderung von Biosicherheitsrisiken, die durch den Missbrauch von künstlicher Intelligenztechnologie entstehen“.Es ist erforderlich, eine umfassende Überwachung der gesamten Lieferkette für Technologien wie die Genomeditierung und die synthetische Biologie zu implementieren.Setzen Sie sichere Grenzen für die technologische Entwicklung.
Derzeit verfügt China in den Bereichen diskrete Diffusionsmodelle und biologisches Sequenzdesign über eine vollständige Innovationskette aus „Theorie-Anwendung-Einrichtungen-Standards“. Diese Fortschritte werden nicht nur die grundlegende Logik der biomedizinischen Forschung und Entwicklung neu gestalten, sondern wahrscheinlich auch eine Revolution in der Biotechnologiebranche einer neuen Generation auslösen. Die saudische Medienzeitung Mecca schrieb dazu: „China holt nicht nur gegenüber dem Westen auf, sondern etabliert auch seine eigenen innovativen Merkmale. Die jüngere Generation von Innovatoren konzentriert sich auf Spitzentechnologien, was China zu einer weltweit führenden Kraft in der Biotechnologie macht und voraussichtlich zu einer globalen Biotechnologiemacht werden lässt.“
Quellen:
1.https://export.shobserver.com/baijiahao/html/709277.html
2.https://www.ncsti.gov.cn/kjdt/yqdy/cpy2/zchj/202410/t20241012_181850.html
3.https://sghexport.shobserver.com/html/baijiahao/2023/06/15/1051928.html
4.http://news.china.com.cn/2025-01/03/content_117643069.shtml