Command Palette
Search for a command to run...
Ausgewählt Für ICML! MIT-Team Erzielt Neuen Durchbruch Auf Basis Von AlphaFold Und Enthüllt Die Dynamische Vielfalt Von Proteinen
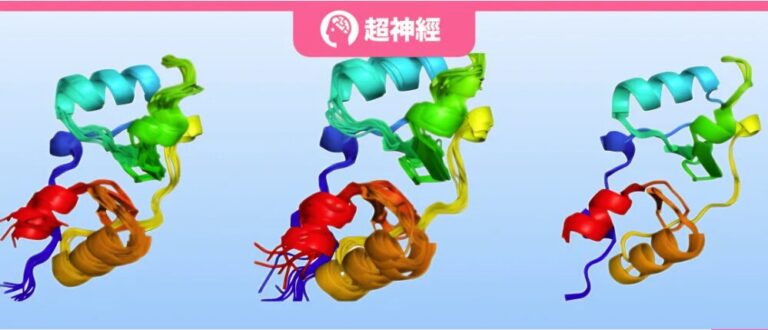
Als wichtiger Bestandteil von Organismen haben Proteine unterschiedliche Zustände und nehmen komplexe dreidimensionale Strukturen an, die auf unterschiedlichen Strukturkombinationen kollektiver Bewegungen oder ungeordneter Fluktuationen basieren, um vielfältige biologische Funktionen zu erfüllen. Beispielsweise sind Proteinkonformationsänderungen für die Funktionen von Transportern, Kanälen und Enzymen von entscheidender Bedeutung, während die Eigenschaften der ausgewogenen Kombination dazu beitragen, die Stärke und Selektivität molekularer Interaktionen zu steuern.
In den letzten Jahren haben Deep-Learning-Methoden wie AlphaFold große Erfolge bei der Einzelzustandsmodellierung von Proteinen erzielt, können jedoch die Konformationsheterogenität nicht erklären. Für Strukturbiologen ist es alsoWie kann eine genaue Vorhersage einer einzelnen Struktur sichergestellt und gleichzeitig mögliche Strukturkombinationen aufgedeckt werden?Es handelt sich um ein schwieriges Problem, das dringend gelöst werden muss.
Kürzlich kombinierte ein Forschungsteam des MIT die neuartigen Sampling-Methoden von AlphaFold und ESMFold und lieferte so eine neue Perspektive zur Beobachtung und zum Verständnis des Konformationsraums von Proteinen durch die Flow-Matching-Technologie.
Diese Studie demonstriert die Leistung der Flow-Matching-Varianten AlphaFlow und ESMFlow in zwei verschiedenen Szenarien.Das Modell wurde schließlich auf der PDB feinabgestimmt und weiter auf dem ATLAS-Datensatz trainiert. Beide zeigten eine überlegene Leistung und übertrafen nicht nur die traditionelle MSA-Basislinie bei der Vorhersage der Konformationsflexibilität und der Modellierung der Atompositionsverteilung, sondern machten auch erhebliche Fortschritte bei der Replikation von Beobachtungen höherer Ordnungsgruppen.
Die zugehörige Forschung mit dem Titel „AlphaFold Meets Flow Matching for Generating Protein Ensembles“ wurde für ICML 2024 ausgewählt, die wichtigste akademische Konferenz im Bereich KI.

Papieradresse:
https://openreview.net/forum?id=rs8Sh2UASt
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Basierend auf PDB- und ATLAS-Datensätzen, um die Fairness der experimentellen Ergebnisse zu gewährleisten
Wie wir alle wissen, wurde AlphaFold durchgängig basierend auf Strukturen im PDB entwickelt und trainiert, während ESMFold Einbettungen aus dem Protein Language Model (PLM) als Eingabe verwendete. daher,In dieser Studie wurden hauptsächlich PDB-Datensätze und MD-Datensätze verwendet.
Um zunächst einen Testsatz strukturell heterogener Proteine aus der PDB zu erstellen, verwendeten wir die SIFTS-Annotationsdatenbank und ihre Zuordnung auf Restebene von PDB-Ketten zu UniProt-Referenzsequenzen und ordneten jede hinterlegte Kette einem Fragment zu. Anschließend wurden alle Fragmente der Cluster basierend auf einem Jaccard-Ähnlichkeitsschwellenwert von 0,75 vollständig verbunden, wobei jeder resultierende Cluster als einzigartiges Protein behandelt wurde.Das Ergebnis waren 75.000 Proteine.
Darüber hinaus wurden in der Studie folgende Daten erhoben:
* Proteine, die vor Ablauf der AlphaFold-Trainingsfrist keine Ketten übermittelt haben, nach Ablauf der Frist jedoch 2–30 Ketten hinterlegt haben;
* Proteine mit Längen zwischen 256–768 Resten;
* Proteine mit mindestens 2 Strukturclustern, wenn der Schwellenwert für die Kettenclusterung 0,85 symmetrisches lDDT-Cα und vollständige Konnektivität betrug.
Schließlich wurden 563 Proteine erhalten, die durch 2.843 Ketten repräsentiert werden.Die Forscher extrahierten 100 Proteine, die durch 500 Ketten repräsentiert wurden, um einen Testsatz zu bilden.
Zweitens erstellten die Forscher den ATLAS-Datensatz auf der Grundlage des MD-Datensatzes.Letztere bestand aus 1.390 Proteinen, die auf Grundlage der ECOD-Domänenklassifizierung ausgewählt wurden.Für jedes Protein bietet der Datensatz drei wiederholte Simulationen mit einer Länge von 100 ns, wobei jede Simulation 10.000 Frames enthält. Um diese Trajektorien zu trainieren und zu validieren, haben wir zunächst mithilfe der bereitgestellten Sequenzen und der ColabFold MMSeqs2-Pipeline MSAs für alle 1.390 ATLAS-Einträge generiert.
Anschließend wählten die Forscher zufällig 300 Konformationen aus der Trainingspipeline aus, wobei sie den 1. Mai 2018 und den 1. Mai 2019 als Trainings- bzw. Validierungstermine verwendeten, und erhielten schließlich 1265/39/82 Trainings-, Validierungs- und Testsätze.

Modellerstellung: Verwenden von AlphaFold als Rauschunterdrückungsmodell zur Durchführung von Flussanpassungen an Proteinsammlungen
Angesichts der erheblichen Herausforderungen bei der Neuentwicklung eines Verteilungsmodells mit der gleichen Genauigkeit und den gleichen Generalisierungsfähigkeiten wie AlphaFold nutzt diese Studie die jüngsten konzeptionellen Fortschritte bei generativen Modellen.Es ist fast unkompliziert, AlphaFold als generatives Modell wiederzuverwenden.

Bislang modellieren typische Diffusionsmodellarchitekturen von Text zu Bildern fast alle die bedingte Verteilung p(x | s) eines Bildes x, das von einem Texthinweis s abhängig ist. Der Kern dieser Modelle ist ein neuronales Netzwerk zur Rauschunterdrückung, das ein verrauschtes Bild und eine Textaufforderung aufnimmt und ein sauberes Bild vorhersagt.
Basierend auf diesen Bedingungen werden solche Modelle normalerweise mit einem einfachen mittleren quadratischen Fehler (MSE) trainiert. In ähnlicher Weise kann ein Proteinstrukturprädiktor, der mit einer regressionsähnlichen Verlustfunktion wie AlphaFold oder ESMFold trainiert wurde, durch die einfache Bereitstellung zusätzlicher verrauschter Struktureingaben in ein rauschfreies Modell umgewandelt werden. Mit diesen architektonischen Anpassungen kann diese Studie AlphaFold und ESMFold weiter in jedes iterative, auf Rauschunterdrückung basierende Framework für generative Modellierung einfügen.
Diese Studie geht davon aus, dass der Entwurf des Frameworks zur Generierung von Flussanpassungen der Auswahl eines bedingten Wahrscheinlichkeitspfads pt(x | x1) und des entsprechenden Vektorfelds ut(x | x1) entspricht. Daher definiert diese Studie ein neu parametrisiertes neuronales Netzwerk x1(x, t; θ), indem es Rauschen x0 von q(x0) abtastet und es linear mit dem Datenpunkt x1 interpoliert, um den bedingten Wahrscheinlichkeitspfad zu definieren.Daher wird die AlphaFold-Architektur als Rauschunterdrückungsmodell verwendet.
Um die Flussanpassung auf Proteinstrukturen anzuwenden, beschreibt die Studie die Struktur auch durch die 3D-Koordinaten ihrer β-Kohlenstoffe (α-Kohlenstoff für Glycin): x ∈ R^N×3. Dadurch wird auch sichergestellt, dass die Eingabe für das neuronale Netzwerk immer eine polymerähnliche, physikalisch plausible dreidimensionale Struktur ist.
Da das Flow-Matching-Framework das Definieren und Umkehren von Rauschprozessen beinhaltet, weist es viele Ähnlichkeiten mit der harmonischen Diffusion von Proteinstrukturen auf, die beide zur gleichen vorherigen Verteilung konvergieren. Als allgemeinerer Rahmen gilt jedochStream Matching bietet zwei Hauptvorteile:
Erste,Die harmonische Diffusion konvergiert nur im zeitlichen Grenzfall unendlich zur vorherigen Verteilung, und die Konvergenzrate hängt von der Datendimensionalität, d. h. der Proteingröße, ab. Dies führt zu einer Verteilungsverschiebung zum Zeitpunkt der Inferenz, wenn das Training nur mit relativ kleinen Pflanzen erfolgt.
Zweitens,Flow Matching bietet eine einfache Möglichkeit, sehr häufige fehlende Rückstände im PDB zu behandeln, indem sie einfach weggelassen werden. Im Gegensatz dazu entstehen bei der harmonischen Diffusion Abhängigkeiten zwischen Atompositionen, was eine Dateninterpolation für fehlende Reste erforderlich macht.

Schließlich wurden im Rahmen der Studie alle Gewichte von AlphaFold und ESMFold auf PDB basierend auf dem Prozessabgleichsrahmen fein abgestimmt, und die verwendeten Trainingsfristen für AlphaFold und ESMFold waren der 1. Mai 2018 bzw. der 1. Mai 2020. Am Ende dieser Trainingsphase erhielt die Studie Flow-Matching-Varianten von AlphaFold und ESMFold,Und nannte es AlphaFLOW und ESMFLOW.
Um die Fähigkeit zu bewerten, aus MD-Ensembles zu lernen, wurden die beiden Modelle anhand des ATLAS-Datensatzes, der MD-Simulationen aller Atome enthält, weiter optimiert. Nach dem Training mit 43.000 bzw. 27.000 zusätzlichen BeispielenDie Studie erhielt MD-spezifische Modellvarianten – AlphaFLOW-MD und ESMFLOW-MD.
Experimentelle Ergebnisse: Die Leistung übertrifft die traditioneller Methoden und bietet breite Anwendungsaussichten im Bereich der Strukturbiologie
Die Forscher bewerteten zunächst die Fähigkeiten von AlphaFLOW und ESMFLOW für verschiedene Konformationen von im PDB abgelegten Proteinen.
Zu diesem Zweck wurde im Rahmen der Studie ein Testset mit 100 Proteinen erstellt, die Anzeichen für mehrere Ketten und konformationelle Heterogenität aufwiesen und nach dem AlphaFold-Trainingstermin (1. Mai 2018) hinterlegt wurden. Anschließend wurden sie anhand von drei Hauptindikatoren bewertet: Präzision, Rückruf und Diversität.

Die Ergebnisse zeigen, dass AlphaFLOW dem MSA-Subsampling insofern ähnelt, als dass beide die Vielfalt der Vorhersagen auf Kosten der Genauigkeit erhöhen. Im Vergleich zum MSA-Subsampling verfolgen AlphaFLOW-Varianten jedoch deutlich bessere Pareto-Fronten.
In Bezug auf Präzision und RückrufAlphaFLOW weist ein sehr ähnliches Verhalten wie MSA-Subsampling auf.Etwas überraschend ist, dass keiner der Ansätze die Gesamterinnerung im Vergleich zum Basis-AlphaFold signifikant verbessert.
Insgesamt ist die Genauigkeit von ESMFold und ESMFLOW im Vergleich zur AlphaFold-Methodenfamilie relativ geringer. Allerdings kann ESMFLOW im Vergleich zum Basismodell ESMFold eine große Vielfalt einbringen.Und verbessern Sie die Rückrufrate, fast ohne Einbußen bei der Präzision.
Darüber hinaus zeigte die RMWD-Analyse dieser Studie, dass AlphaFlow bei der Vorhersage der durchschnittlichen Position von Atomen etwas besser war als AlphaFold und hinsichtlich der Modellvarianz deutlich besser als MSA-Subsampling.

In dieser Studie wurde außerdem die Fähigkeit von AlphaFLOW und ESMFLOW bewertet, Proxy-MD-Ensembles für einen Testsatz von 82 Proteinen in der ATLAS-Datenbank zu generieren. In dieser Studie wurden einzelne Proben aus jeder Methode verwendet und die Ähnlichkeit der Probe mit der MD-Population anhand einer Reihe von Bewertungen untersucht.
Die Ergebnisse zeigen, dassAlphaFLOW-MD erzielt erhebliche Verbesserungen der Ähnlichkeit und übertrifft die Leistung der MSA-Unterabtastung bei weitem.

Da der MD als der wahre Wert angesehen wird, ist die Ausführung bis zur Konvergenz kostspielig. Daher analysiert diese Studie weiter, ob AlphaFLOW bei einem gleichwertigen begrenzten Rechenbudget, z. B. in GPU-Stunden, bessere Ergebnisse liefern kann. Zu diesem Zweck reduzierte die Studie die Anzahl der aus AlphaFLOW entnommenen Proben (von 250 auf 4) und verkürzte die Länge der MD-Trajektorie (von 100 ns auf 160 ps).
Die Ergebnisse zeigen, dass die Qualität des AlphaFLOW-Ensembles konstant bleibt, die MD-Trajektorien jedoch länger brauchen, um das gleiche Qualitätsniveau zu erreichen oder zu überschreiten.
Drei wichtige allgemeine Protein-Vortrainingsmodelle fallen auf, und das Gebiet der Strukturbiologie ist voller Vitalität
In den letzten Jahren kam es immer wieder zu Kollisionen zwischen Proteinen und künstlicher Intelligenz, die neue Funken entfachten.Derzeit hat das universelle Vortraining von Proteinen eine neue Situation mit drei Säulen gebildet.Das sind die DeepMind Alphafold-Reihe, David Bakers RoseTTAFold-Reihe und die Meta ESM-Reihe. Auf der Grundlage dieser drei Modelle hat die Zahl der relevanten wissenschaftlichen Forschungsergebnisse explosionsartig zugenommen. Allein im ersten Halbjahr 2024 wurden mehrere Forschungsergebnisse in Top-Journalen wie Nature und Science veröffentlicht.
Im März 2024 veröffentlichten Forscher der University of North Carolina School of Medicine, der University of California, San Francisco, der Stanford University und der Harvard University eine Studie in Science, die bestätigte, dassDie vorhergesagten Strukturen von AlphaFold2 können als Leitfaden für die zukünftige Arzneimittelentdeckung dienen.Das Forschungsteam stellte fest, dass AlphaFold2 erhebliche praktische Anwendungsmöglichkeiten in den Bereichen Strukturbiologie, Proteindesign, Interaktionen, Zielvorhersage, Funktionsvorhersage und biologische Mechanismen zeigte und in der Lage war, durch das Screening von Milliarden von Verbindungen und den Abgleich von Bibliotheken mit Proteinstrukturen nach potenziellen neuen Medikamenten zu suchen.
Im Mai 2024 veröffentlichte das Google DeepMind-Team AlphaFold 3 in Nature und erweiterte damit die Technologie über die Proteinfaltung hinaus, um die Struktur und Wechselwirkungen von Lebensmolekülen wie Proteinen, DNA, RNA und Liganden mit beispielloser Präzision vorherzusagen. Das heisst,AlphaFold 3 wird die Arzneimittelentwicklung und Genomforschung weiter beschleunigen.Beginn einer neuen Ära der Zellbiologie mit künstlicher Intelligenz.
Mit der Veröffentlichung von AlphaFold 3Die Alphafold-Serie hat endlich eine rein atomare Grundlage geschaffen.In ähnlicher Weise wurde im ersten Halbjahr dieses Jahres auch RoseTTAFold All-Atom erfolgreich in die RoseTTAFold-Reihe eingeführt, wodurch die Möglichkeit geschaffen wurde, vernünftige Vorhersagen über kovalente Proteinmodifikationen und die Zusammensetzung mehrerer Nukleinsäureketten und kleiner Moleküle zu treffen.
Mithilfe von Alphafold3 und RoseTTAFold All-Atom können Forscher der Vorstellungskraft freien Lauf lassen. So veröffentlichte beispielsweise im Juni 2024 ein internationales Forschungsteam einen Artikel in Nature Biotechnology, in dem es demonstrierte, wie man mit der Strategie der Kombination von AlphaFold 3 und RoseTTAFold All-Atom erfolgreich ein neues Proteingerüst entwickeln kann, das Medikamente effektiver direkt an erkrankte Zellen abgeben und so die therapeutische Wirkung verbessern und Nebenwirkungen reduzieren kann. Diese Entdeckung stellt einen großen Fortschritt bei der Anwendung von KI in der Präzisionsmedizin dar.
Leider löste Meta im August 2023 das ESMFold-Team auf und konzentrierte sich voll und ganz auf die Förderung der Kommerzialisierung von KI. Doch die Forschung an der ESM-Reihe ist nicht beendet. Beispielsweise hat das Modell wichtige Fortschritte im Bereich der Proteinsprachenmodellierung erzielt und eine einheitliche Modellierungslösung bereitgestellt, die Informationen auf mehreren Skalen integriert. Insbesondere handelt es sich dabei um das erste auf Proteinen vortrainierte Sprachmodell, das sowohl Aminosäureinformationen als auch Atominformationen verarbeiten kann.
Daraus lässt sich erkennen, dassIn der neuen Ära, in der die Alphafold-Serie, die RoseTTAFold-Serie und die ESM-Serie Hand in Hand gehen,Die Verbindung von KI und Proteinforschung wird enger, was nicht nur unser Verständnis der Struktur und Funktion von Proteinen beschleunigen, sondern auch revolutionäre Veränderungen in der Krankheitsbehandlung, der Arzneimittelentwicklung und den biotechnologischen Anwendungen mit sich bringen wird. Durch die sprunghafte Entwicklung, die die KI-Technologie mit sich bringt, gewinnt das Gebiet der Strukturbiologie an Dynamik und es beginnt langsam ein neues Kapitel in der Biomedizin.








