Command Palette
Search for a command to run...
Kann Die Vermehrung Von Krebszellen Hemmen! Die Huihu School of Pharmacy Und Die Tianjin Medical University Haben Gemeinsam Einen Neuen Tumorsuppressor-Protein-Degrader dp53m Entwickelt

Vielleicht wissen viele Leute nicht,Tatsächlich hat jeder von uns Krebszellen in seinem Körper.Im menschlichen Körper werden täglich Milliarden oder sogar Zehnmilliarden Zellen gebildet und ersetzt. Während dieses Stoffwechselprozesses kommt es zwangsläufig zu Fehlern bei der DNA-Replikation, beispielsweise zu Genmutationen, die normale Zellen direkt vor Ort in Krebszellen verwandeln. Aber,Es gibt einige Tumorsuppressorproteine im menschlichen Körper.Diese Proteine, wie beispielsweise p53, können die Entstehung von Krebs eindämmen, indem sie den Zellzyklus regulieren und die Apoptose oder Seneszenz von Krebszellen herbeiführen. Dies ist auch ein wichtiger Grund, warum die meisten von uns friedlich mit Krebszellen koexistieren können.
Das Tumorsuppressorprotein p53, bekannt als „Wächter des Genoms“, wird durch das Gen TP53 kodiert und spielt eine wichtige Rolle bei der Verhinderung der Entstehung von Krebs.Allerdings kann es bei TP53 an mehreren häufigen spezifischen Hotspots wie R175, G245, R248, R273 und R282 zu Missense-Mutationen kommen, die zu p53-Mutanten führen und den Verlust der normalen Funktion des Tumorsuppressorproteins zur Folge haben. Darüber hinaus verliert das mutierte p53 aufgrund der dominanten negativen Wirkung einiger p53-Mutanten nicht nur seine ursprüngliche Tumorsuppressorfunktion, sondern beeinträchtigt auch die Tumorsuppressoraktivität des normalen Wildtyp-p53 (p53-WT), wodurch das Risiko der Tumorentstehung steigt.
Im Vergleich zu mehreren anderen p53-MutantenDas mutierte p53-R175H-Protein hat ein höheres Potenzial für Tumorbildung, Metastasierung und Arzneimittelresistenz.Die Entwicklung von Medikamenten, die auf p53-R175H abzielen und die Ermöglichung einer präzisen Identifizierung und Degradation von p53-R175H durch zielgerichtete Medikamente ist eine wirksame Strategie zur Hemmung der Entstehung von Krebs. Da den meisten mutierten p53-Proteinen jedoch aktive Zentren für niedermolekulare Medikamente fehlen, ist es äußerst schwierig, zielgerichtete Medikamente zu entwickeln, die mutierte p53-Proteine genau erkennen und beeinflussen können.
In diesem ZusammenhangProfessor Wu Sijin von der Huihu School of Pharmacy an der Xi'an Jiaotong-Liverpool University, zusammen mit Professor Xie Songbo und Professor Zhong Diansheng vom Tianjin Medical University General Hospital,Ein Artikel mit dem Titel „Ein auf DNA-Aptameren basierendes PROTAC für die präzise Therapie von durch p53-R175H-Hotspot-Mutationen verursachtem Krebs“ wurde in Elsevier veröffentlicht.
In dieser Studie wurde ein selektiver p53-R175H-Degrader, dp53m, entwickelt.Dieser Degrader kann das mutierte p53-R175H-Protein spezifisch erkennen und das natürliche Proteinabbausystem der Zelle – das Ubiquitin-Protease-System – nutzen, um einen gezielten Abbau des Zielproteins zu erreichen und die funktionelle Expression des mutierten p53-Proteins zu hemmen.
Der Degrader weist eine signifikante Antitumorwirkung auf und zeigt keine offensichtlichen toxischen Reaktionen. Darüber hinaus kann dp53m auch synergistisch mit dem Chemotherapeutikum Cisplatin zusammenwirken, um die Empfindlichkeit von Krebszellen gegenüber Cisplatin zu erhöhen, was für die Behandlung von Krebs entscheidend ist.
Forschungshighlights:
- Die Forscher modifizierten Nicht-Kernbasen unter Anleitung von MD-Simulationen und führten das RNA-Aptamer durch iteratives, molekulares Docking-gesteuertes Post-SELEX-Verfahren zu einem DNA-Aptamer (p53m-DA).
- Als Komponente von dp53m kann p53m-DA das Protein p53-R175H spezifisch erkennen. CRBN ist am Proteinabbau beteiligt, sodass dp53m das Protein p53-R175H spezifisch erkennen und abbauen kann.

Papieradresse:
https://pubmed.ncbi.nlm.nih.gov/38811338
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt außerdem umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Post-SELEX-Engineering: Konstruktion eines Hochleistungs-DNA-Aptamers, der spezifisch mutiertes p53-R175H erkennt
Bei der Untersuchung von p53 haben Forscher herausgefunden, dass die R175H-Mutation die DNA-Bindungsfunktion beeinflusst, indem sie den Strukturzustand von p53 verändert. Die Kombination mehrerer kleiner Inhibitormoleküle und Aptamere kann die Aktivität von p53-R175H teilweise wiederherstellen, der molekulare Mechanismus seiner Auswirkung wurde jedoch nicht beschrieben.
In dieser Studie wurden erstmals Methoden der molekularen Simulation eingesetzt, um den intrinsischen molekularen Mechanismus von Aptameren bei der Wiederherstellung der Funktion von p53-R175H zu untersuchen.Im Vergleich zum normalen p53-WT ist beim mutierten p53-R175H der Abstand zwischen L3 und C-Helix geringer und der Abstand zwischen L2 und L3 größer. Die strukturellen Veränderungen an diesen Positionen könnten der Hauptgrund dafür sein, dass der p53-R175H-Mutant seine typische Funktion verliert und bieten zudem eine strukturelle Grundlage für die spezifische Erkennung seiner Aptamere und verwandter PROTAC-Moleküle.
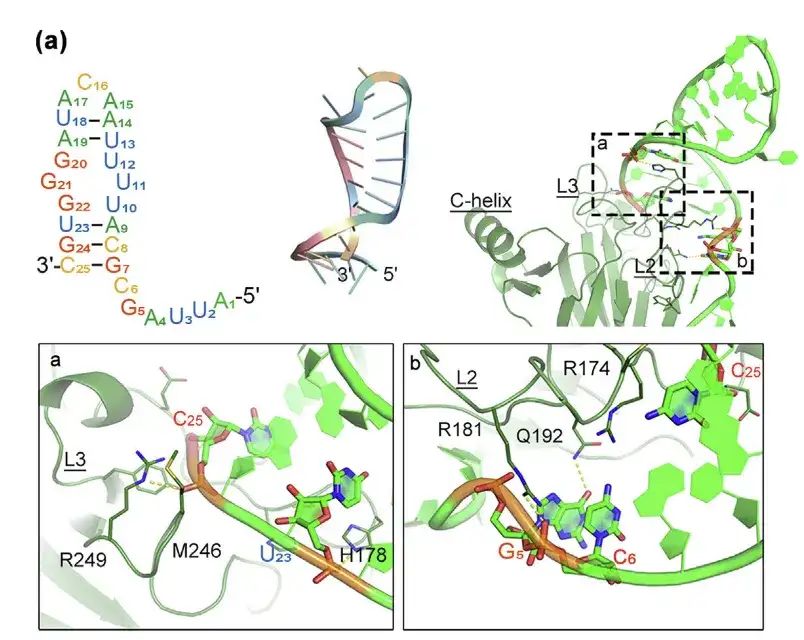
- Vorhersage der Struktur von p53m-RA und seiner Interaktion mit dem p53-R175H-Protein mittels molekularer Modellierung
- Die Verbindungsschnittstelle ist in den Bereichen a und b dargestellt.
In früheren Studien wurde gezeigt, dass das auf p53-R175H abzielende RNA-Aptamer (p53m-RA) eine hohe Affinität zu p53-R175H aufweist. Es ist jedoch im Serum sehr instabil, was seine praktische Anwendung einschränkt.
Quellen:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9884801
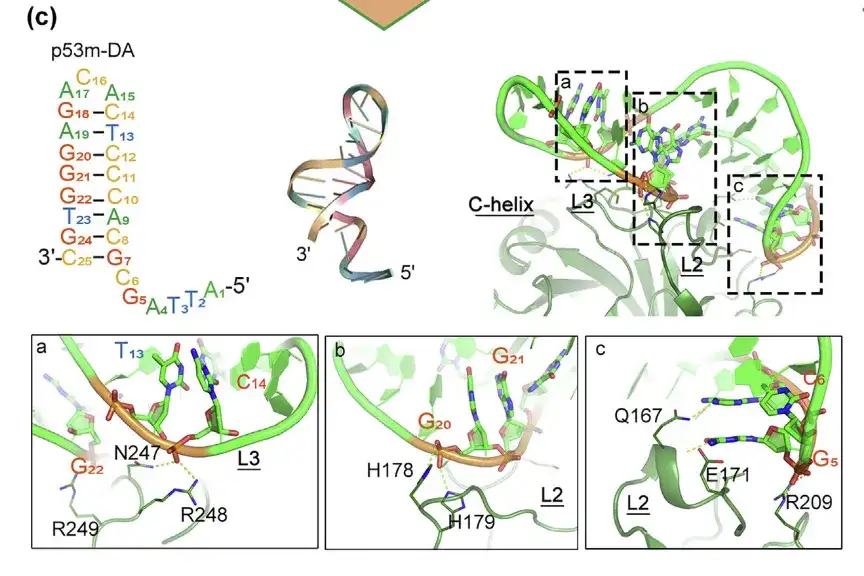
Post-SELEX-optimiertes p53m-DA und seine Interaktion mit p53-R175H
Die Verbindungsschnittstelle befindet sich in den Bereichen a, b und c
Um die strukturelle Stabilität und Affinität des Aptamers zu verbessern, modifizierten die Forscher die Nicht-Kernbasen unter Anleitung einer MD-Simulation und wandelten den RNA-Aptamer durch Post-SELEX in einen DNA-Aptamer (p53m-DA) um.
Die Rechenleistung der Molekülsimulationen in dieser Studie wurde bereitgestellt von OpenBayes liefern
Die Ergebnisse zeigten, dass die Struktur der gepaarten Region von p53m-DA eine große Furche war und die ungepaarte Schleifenregion eine kleine Furche war. Die große Furche spielt eine Schlüsselrolle bei der Bindung mit H178, H179, E171 und Q167 in L2, und die kleine Furche spielt eine Schlüsselrolle bei der Interaktion mit N247, R248 und R249 in L3.Darüber hinaus behielt p53m-DA während der gesamten Simulation kontinuierlich eine doppelsträngige helikale Konformation bei, was auf die Stabilität seiner Struktur hinweist.
L2 und L3 sind Regionen, die im mutierten Protein vorhanden sind
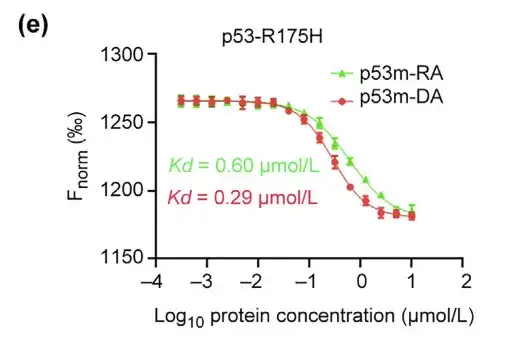
Die Forscher untersuchten außerdem die Affinität und Selektivität von p53m-DA für p53-R175H. Der MST-Test zeigte, dass der Kd-Wert von p53m-DA 0,29 lmol/l betrug und damit etwa doppelt so niedrig war wie der von p53m-RA.
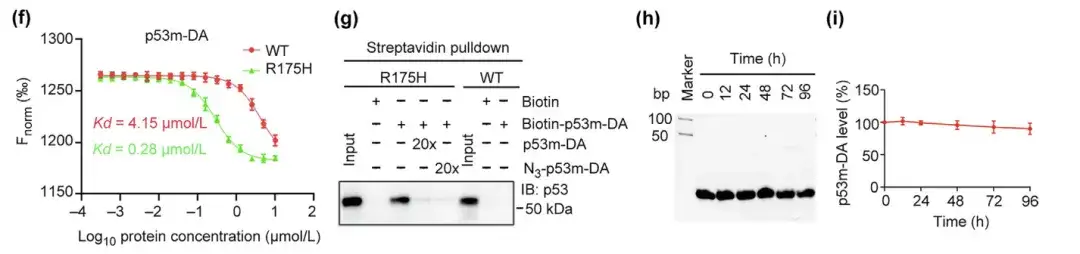
(f) Affinität von p53m-DA für p53-WT und p53-R175H
(g) Spezifische Bindung von p53m-DA an p53-R175H mittels Streptavidin-Pulldown-Assay
(h,i) Bewertung der Serumstabilität von p53m-DA und Bestimmung der Restmenge an p53m-DA
Darüber hinaus war die Affinität von p53m-DA zum mutierten p53-R175H 14-mal höher als seine Affinität zum normalen p53-WT. Im Streptavidin-Pulldown-Test band p53m-DA spezifisch an p53-R175H, interagierte jedoch nicht mit p53-WT, was auf die Spezifität von p53m-DA für p53-R175H hinweist. Bemerkenswerterweise war die Serumstabilität von p53m-DA deutlich verbessert, mit minimaler Degradation nach 96 Stunden.
Zusammenfassend:p53m-DA ist ein Aptamer mit hoher Spezifität und Stabilität.
dp53m: ein spezifischer p53-R175H-Degrader
Experiment 1: Synthese von DP53M
Das 5'-Ende des Aptamers p53m-DA befindet sich außerhalb seiner Bindungsstelle bei p53-R175H. Die Forscher verwendeten eine Klickreaktion, um Alkinylthalidomid (einen Liganden für CRBN E3-Ligase) an das 5'-Ende von N3-p53m-DA zu konjugieren. Die daraus resultierende, auf Proteinabbau abzielende Chimäre PROTAC (genannt dp53m) konnte selektiv an p53-R175H binden. Die dp53m-Struktur ist in der folgenden Abbildung dargestellt:
*PROTAC: besteht aus einem E3-Ligase-Liganden, einem Liganden, der auf POI abzielt, und einem chemischen Linker zwischen den Liganden
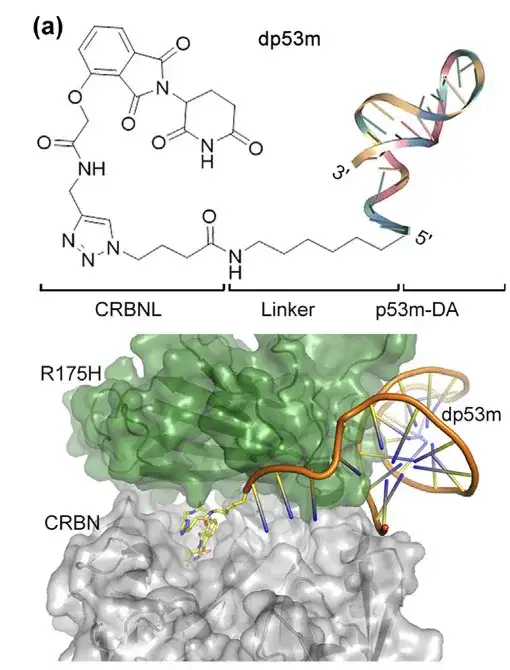
(a) Die Struktur von dp53m wurde mithilfe von Protein-Protein-Docking und molekulardynamischen Berechnungen simuliert.
Unten: Seine Bindungsschnittstelle mit p53-R175H und CRBN
Experiment 2: dp53m kann mutiertes p53-R175H spezifisch erkennen und abbauen
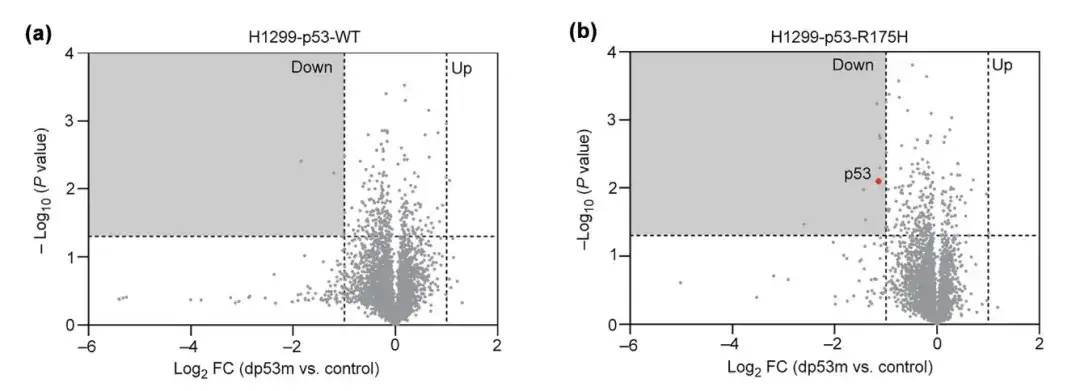
Um die zelluläre Spezifität von dp53m für p53-R175H zu untersuchen, wurden in H1299-Zellen überexprimierte p53-R175H und p53-WT 16 Stunden lang mit PBS oder dp53m behandelt und ihre Proteinänderungsniveaus überwacht. Die Ergebnisse zeigten, dass dp53m p53-R175H signifikant abbaute, nicht jedoch p53-WT.
*PBS ist eine phosphatgepufferte Kochsalzlösung, die als Kontrollgruppe verwendet wird
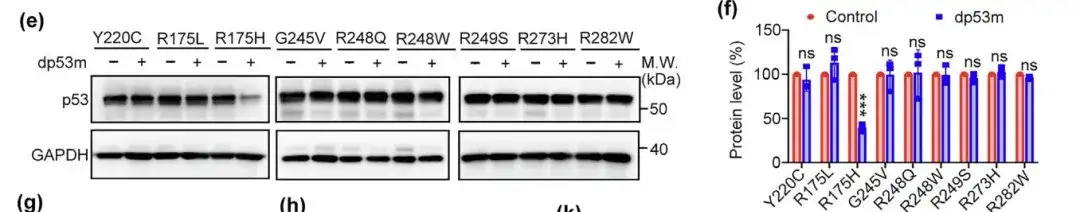
Darüber hinaus bewerteten die Forscher auch das Abbaupotenzial von dp53m gegenüber anderen Hotspot-Mutanten wie Y220C, R175L, G245V, R248Q, R248W, R249S, R273H und R282W. Die Ergebnisse zeigten, dass dp53m nur p53-R175H effektiv abbaute.Seine Spezifität für den Abbau von p53-R175H wurde nachgewiesen.
Im Vergleich zu p53WT erhöhte die Behandlung mit dp53m die Polyubiquitinierung von p53-R175H. Studien haben gezeigt, dass der Abbau von dp53m durch den Proteaseinhibitor MG132, den CRBN-Liganden oder CRBN-gerichtete siRNA blockiert werden kann.Es wird vermutet, dass der durch dp53m induzierte Abbau von p53-R175H über den Ubiquitin-Proteasom-Mechanismus erfolgt.
Zusammenfassend:dp53m ist ein spezifischer p53-R175H-Degrader und sein Abbauprozess erfolgt über den Ubiquitin-Proteasom-Mechanismus.
dp53m hemmt die Proliferation und Migration von Krebszellen sowie das Tumorwachstum
Experiment 1: dp53m hemmt die durch p53-R175H induzierte Krebszellproliferation und -migration
Die Forscher behandelten H1299-p53-WT-, H1299-p53-R175H-, A549-, Detroit 562- und SKBR3-Zellen mit PBS, p53m-DA bzw. dp53 m, führten Lebensfähigkeitstests an ihnen durch und beobachteten die Koloniebildung in den Zellen.
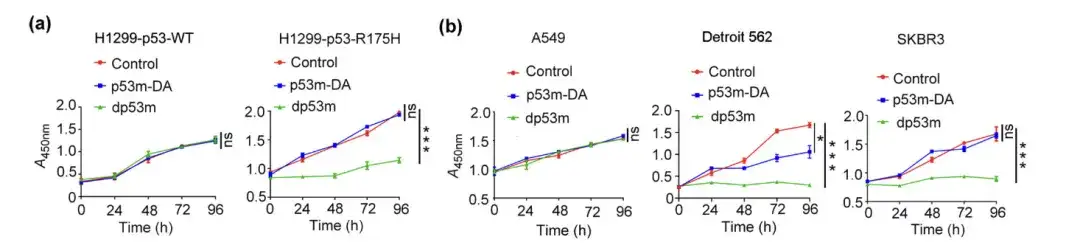
Lebensfähigkeitstest von H1299-p53-WT-, H1299-p53-R175H-, A549-, Detroit 562- und SKBR3-Zellen, die mit PBS, p53m-DA oder dp53m behandelt wurden
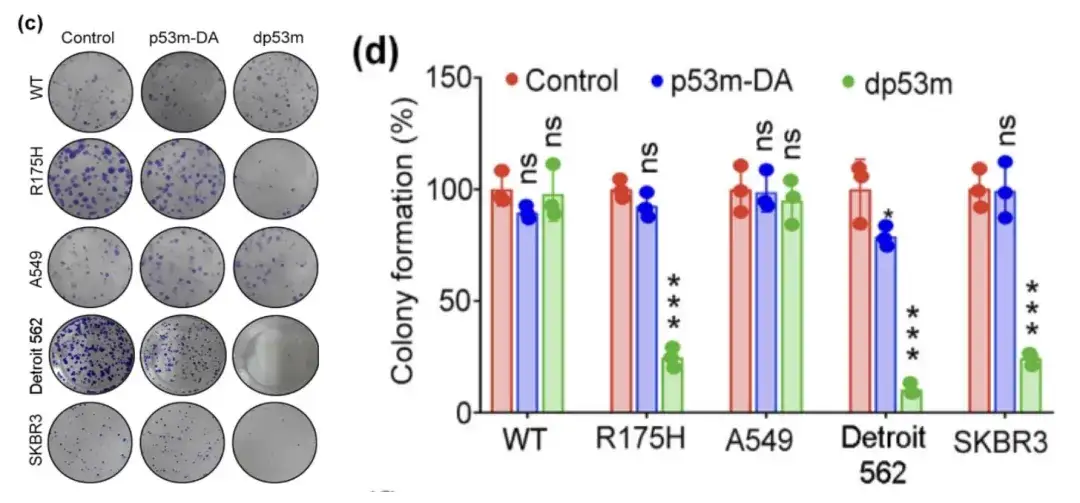
* dp53m hemmt die Koloniebildung in H1299-p53-R175H-, Detroit 562- und SKBR3-Zellen
* In H1299-p53-WT- und A549-Zellen wurde kein signifikanter Effekt beobachtet
Die obige Abbildung zeigt, dass dp53m eine signifikante proliferationshemmende Wirkung auf Krebszellen hat, die p53-R175H (H1299-p53-R175H, Detroit 562 und SKBR3) exprimieren, während Zellen, die p53-WT exprimieren, grundsätzlich nicht betroffen sind.Dies deutet darauf hin, dass dp53m ein großes therapeutisches Potenzial bei Krebszellen mit der Mutation p53-R175H hat.
Experiment 2: dp53m hemmt das Wachstum von p53-R175H-induzierten Tumoren in vivo
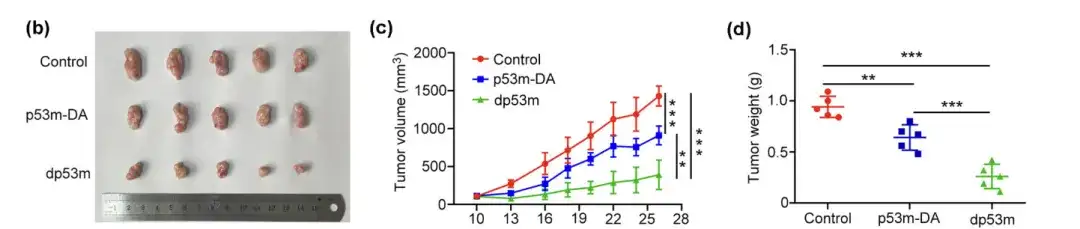
(b) Makroskopisches Erscheinungsbild isolierter Tumoren nach der Behandlung
(c) Zum angegebenen Zeitpunkt gemessenes Tumorvolumen
(d) Bestimmung des endgültigen Tumorgewichts
Die Forscher teilten tumortragende BALB/c-Mäuse nach dem Zufallsprinzip drei Gruppen zu und verabreichten ihnen intravenös p53m-DA, dp53m oder Kochsalzlösung als Kontrolle. Im Vergleich zur Kontrollgruppe oder p53m-DA,dp53m hemmte das Tumorwachstum signifikant.
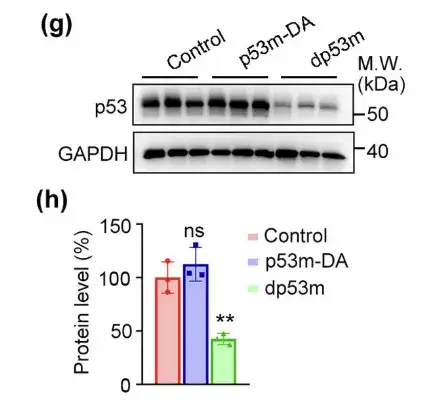
Wie erwartet,dp53m reduzierte effektiv den Expressionsgrad von p53-R175H in Tumoren.
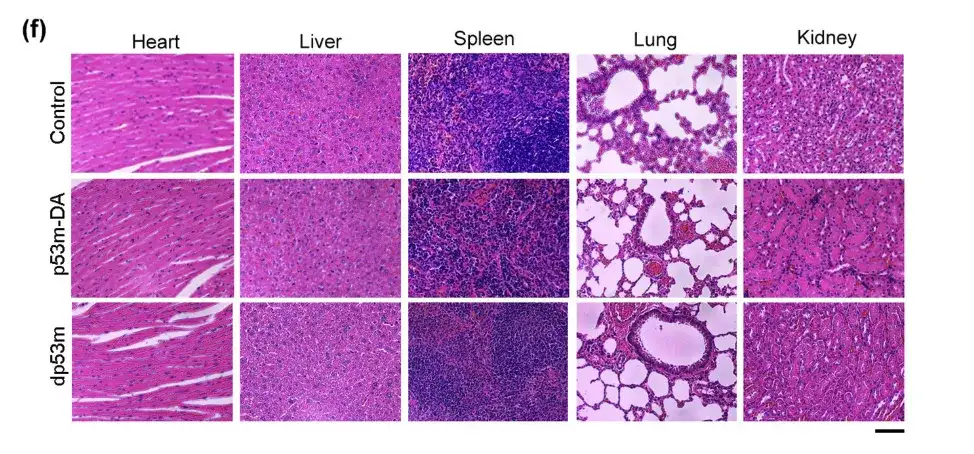
In der oberen Abbildung gab es keine signifikanten Unterschiede in der Histologie des Herz-, Leber-, Lungen-, Milz- und Nierengewebes zwischen den drei Mäusegruppen.Dies lässt darauf schließen, dass dp53m in vivo keine Toxizität hervorruft.
Zusammenfassend:dp53m kann das Wachstum von p53-R175H-induzierten Tumoren ohne offensichtliche Toxizität hemmen.
Da die Mutation p53-R175H mit einer Resistenz gegen Chemotherapeutika wie Cisplatin in Zusammenhang steht, analysierten die Forscher außerdem die Wirkung von dp53m auf die Antitumoraktivität von Cisplatin. Die Ergebnisse zeigten, dass eine starke synergistische Wechselwirkung zwischen dp53m und Cisplatin bestand.Das heißt, dp53m kann die Empfindlichkeit von p53-R175H-Krebszellen gegenüber Cisplatin erhöhen.Damit steht ein „unbesiegbarer Partner“ für die Krebsbehandlung zur Verfügung.
Wissenschaftler stärken die Abwehrkräfte gegen Krebs und bauen gemeinsam einen Weg der Hoffnung
Krebs ist eine Krankheit, die durch unkontrolliertes Zellwachstum verursacht wird. Aufgrund der Alterung der Weltbevölkerung, der zunehmenden Umweltverschmutzung und der veränderten Lebensgewohnheiten hat die Häufigkeit dieser Erkrankung erheblich zugenommen. Nach Angaben der Internationalen Agentur für Krebsforschung (IARC) wird die Zahl der weltweiten Krebsneuerkrankungen voraussichtlich von etwa 19,29 Millionen Fällen im Jahr 2020 auf etwa 30,23 Millionen Fälle im Jahr 2040 ansteigen, was einem Anstieg von 56,71 % entspricht.
Forscher spielen im weltweiten Kampf gegen den Krebs eine entscheidende Rolle. Die 3 korrespondierenden Autoren dieser Studie sind:Herr Wu Sijin von der Huihu School of Pharmacy der Xi'an Jiaotong-Liverpool University, Professor Xie Songbo und Professor Zhong Diansheng vom Tianjin Medical University General Hospital sind herausragende Vertreter auf diesem Gebiet.
Unter ihnen konzentriert sich Professor Wu Sijin auf die Forschung und Entwicklung neuer zielgerichteter Medikamente. Sein aktueller Forschungsschwerpunkt liegt auf der Nutzung computergestützter Arzneimittelentwicklung und -entdeckung zur Identifizierung neuer therapeutischer Arzneimittel unter Einsatz von Simulationsmethoden wie Modellierung, Pharmakophoranalyse, Docking und Molekulardynamik (MD). In seiner Studie aus dem Jahr 2022 stellte er fest, dass eine hochregulierte SOST-Expression mit einer schlechten Prognose bei Brustkrebspatientinnen verbunden war und SOST nachgelagerte Signalwege aktivierte, um die Proliferation von Brustkrebszellen und die Bildung von Knochenmetastasen zu fördern. Durch computergestütztes Screening wurde im Rahmen der Studie ein therapeutischer Wirkstoffkandidat namens S6 identifiziert, der die Interaktion zwischen SOST und STAT3 unterbrechen kann, um die STAT3-Phosphorylierung zu hemmen und die Knochenmetastasierung bei Brustkrebs zu verringern.
Papieradresse:
https://pubmed.ncbi.nlm.nih.gov/36581888
Darüber hinaus schlug Professor Xie Songbo, dessen Forschungsschwerpunkt der gezielte Proteinabbau und die Arzneimittelverabreichung ist, in seiner Studie aus dem Jahr 2023 vor, dass eine neue Strategie verwendet werden könne, um den Abbau „nicht medikamentös behandelbarer“ Proteine herbeizuführen, indem Aptamere als „gezielte Sprengköpfe“ eingesetzt werden. Um dieses Konzept zu bestätigen, wählten die Forscher onkogenes Nukleolin (NCL) als Ziel und erzeugten schließlich eine Reihe von NCL-Degradern. dNCL#T1 induzierte den NCL-Abbau auf eine Weise, die vom Ubiquitin-Protease-System abhing, was die durch NCL induzierte Proliferation von Brustkrebszellen hemmen konnte. Diese Studie eröffnete nicht nur eine neue Perspektive für den Proteinabbau, sondern legte auch eine solide Grundlage für die Entwicklung therapeutischer Medikamente gegen Krankheiten wie Krebs.
Papieradresse:
https://pubmed.ncbi.nlm.nih.gov/36608275
Schließlich hat Professor Zhong Diansheng auch in den Bereichen Frühdiagnose, Chemotherapie, zielgerichtete Therapie und antiangiogene Therapie von Lungenkrebs eingehende Forschungen durchgeführt und mehr als 90 wissenschaftliche Arbeiten veröffentlicht. In seiner Studie aus dem Jahr 2024 stellte er fest, dass CBX4 die Proliferation von Lungenadenokarzinomen (LUAD) durch Hochregulierung der PHGDH-Expression und Serinbiosynthese förderte und gleichzeitig die LUAD-Metastasierung durch Unterdrückung der ZEB2-Transkription hemmte. Dieser Befund hilft uns, die Interaktion zwischen CBX4 und epigenetischen Faktoren zu verstehen und bietet Einblicke in mögliche Therapieansätze für LUAD.
Papieradresse:
https://www.nature.com/articles/s41419-024-06745-z
Neben Lehrer Wu Sijin, Professor Xie Songbo und Professor Zhong Diansheng gibt es viele Wissenschaftler und Ärzte, die sich still und leise engagieren und hart arbeiten. Wir freuen uns auf den Tag, an dem Krebs aufgrund der fortschreitenden Fortschritte in Wissenschaft und Technologie kein Albtraum mehr für die Menschheit sein wird.








