Command Palette
Search for a command to run...
Die Shandong University Entwickelt Einen Interpretierbaren Deep-Learning-Algorithmus Namens RetroExplainer, Um Den Retrosyntheseweg Organischer Verbindungen in 4 Schritten Zu Identifizieren

Ziel der Retrosynthese ist es, eine Reihe geeigneter Reaktanten zu finden, um das Zielprodukt effizient zu synthetisieren. Dies ist eine wichtige Methode zur Lösung organischer Synthesewege und zugleich die einfachste und grundlegendste Methode zum Entwerfen organischer Synthesewege.
Die frühe retrosynthetische Forschung stützte sich größtenteils auf Programmierung, doch später wurde diese Arbeit von der KI übernommen. Allerdings konzentrieren sich bestehende Retrosynthesemethoden meist auf die einstufige Retrosynthese, sind schlecht interpretierbar und können sowohl die Nah- als auch die Fernbereichsinformationen von Molekülen nicht berücksichtigen, was zu einer eingeschränkten Leistung führt.
Zu diesem Zweck haben Wei Leyi von der Shandong-Universität und die Forschungsgruppe von Zou Quan von der University of Electronic Science and Technology of China gemeinsam RetroExplainer entwickelt. Dieser erklärbare Deep-Learning-Algorithmus kann die Retrosynthesewege organischer Verbindungen in 4 Schritten identifizieren und leicht verfügbare Reaktanten bereitstellen. RetroExplainer soll ein leistungsfähiges Werkzeug für die retrosynthetische Forschung in der organischen Chemie darstellen.
Autor | Xuecai
Herausgeber | Sanyang
Ziel der Retrosynthese in der organischen Chemie ist es, eine Reihe geeigneter Reaktanten zu finden, um das Zielprodukt effizient zu synthetisieren.. Dieses Verfahren ist eine unverzichtbare Grundlagenarbeit in der computergestützten Synthese.
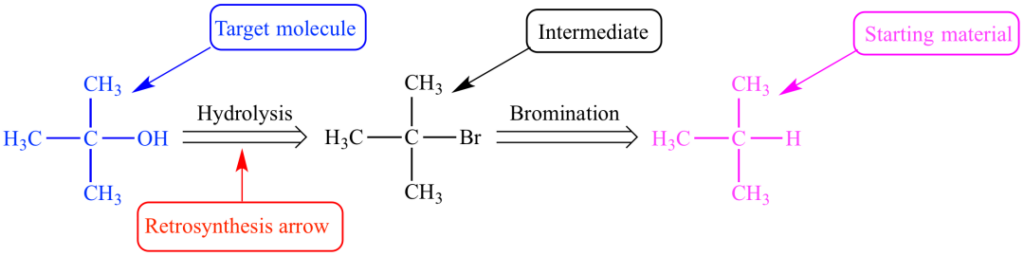
Abbildung 1: Retrosyntheseweg von tert-Butylalkohol
In den 1960er JahrenCorey et al. versuchte, eine retrosynthetische Analyse mittels Programmierung durchzuführenund entwickelte die Software Organic Chemical Simulation Synthesis (OCSS). Jedoch,Mit zunehmender Datenmenge wird diese Arbeit schnell von der KI übernommen. Darunter wird das Deep-Learning-Modell mit großer Spannung erwartet und hat beachtliche Ergebnisse hervorgebracht.
In der frühen KI-Retrosyntheseforschung arbeiteten die Forscher häufig rückwärts von Produkten zu Reaktanten, basierend auf Reaktionsvorlagen, d. h. einer vorlagenbasierten Retrosynthese.. Unter ihnen werden molekulare Fingerabdrücke auf der Basis mehrschichtiger Perzeptronen häufig zur Produktkodierung und Vorlagenauswahl verwendet.
Dann,Forscher begannen, vorlagenfreie und semi-vorlagenbasierte Synthesemethoden zu erforschen, hauptsächlich einschließlich:
1. Sequenzbasierte Retrosynthese;
2. Diagrammbasierte Retrosynthese.
Der Hauptunterschied zwischen beiden liegt in der Form, in der die Moleküle ausgedrückt werden. Ersteres verwendet linearisierte Zeichenfolgen zur Darstellung von Molekülen, wie beispielsweise die SMILES-Spezifikation; während letztere molekulare Graphmodelle zur Darstellung von Molekülen verwendet, hauptsächlich einschließlich der Vorhersage von Reaktionszentren (RC) und der Vervollständigung von Synthonen.
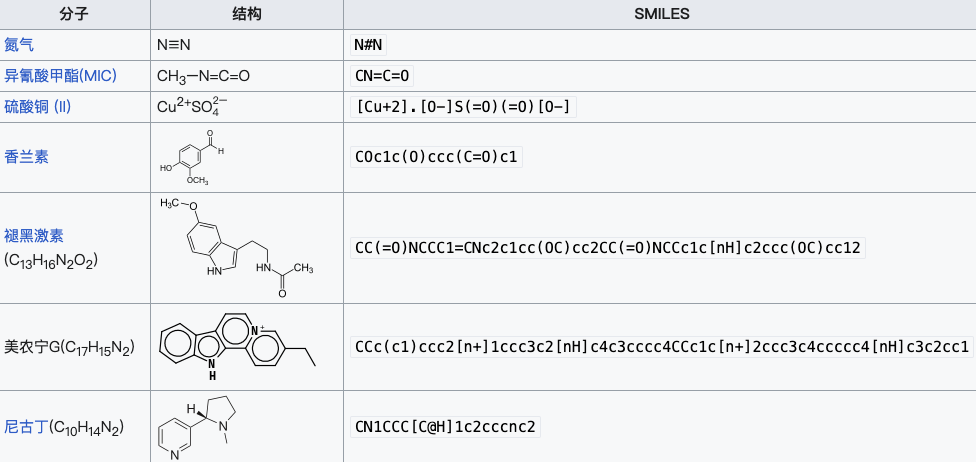
Abbildung 2: SMILES-Ausdrücke einiger Substanzen
Obwohl die bestehenden Retrosynthesemethoden erhebliche Fortschritte gemacht haben,Es gibt jedoch noch drei endogene Probleme:
1.Bei der sequenzbasierten Retrosynthese werden molekulare Informationen übersehen, während bei der graphenbasierten Retrosynthese Sequenzinformationen und weitreichende Merkmale von Molekülen ignoriert werden.. Beide Methoden sind beim Merkmalslernen eingeschränkt und es ist schwierig, die Leistung zu verbessern.
2.Deep Learning-basierte Retrosynthesemethoden weisen eine schlechte Interpretierbarkeit auf. Obwohl die vorlagenbasierte Retrosynthese einen verständlichen Syntheseweg bieten kann, ist der Entscheidungsmechanismus des Algorithmus noch vage und die Reproduzierbarkeit und Durchführbarkeit des Modells müssen berücksichtigt werden.
3.Bestehende Methoden konzentrieren sich meist auf die einstufige Retrosynthese. Dieser Ansatz scheint brauchbare Reaktanten zu ergeben, diese Reaktanten sind jedoch möglicherweise schwer zu beschaffen oder erfordern eine komplizierte Aufbereitung. Daher kann eine mehrstufige Retrosynthese in der tatsächlichen chemischen Synthese sinnvoller sein.
zu diesem Zweck,Wei Leyi von der Shandong University und die Forschungsgruppe von Zou Quan von der University of Electronic Science and Technology of China haben gemeinsam RetroExplainer entwickelt . Dieser Algorithmus kann retrosynthetische Vorhersagen auf der Grundlage von Deep Learning durchführen und dabei die Interpretierbarkeit und Durchführbarkeit des Algorithmus berücksichtigen. RetroExplainer übertrifft andere Algorithmen in fast 12 Benchmark-Datensätzen und 86,91 % der Reaktionen in den vorgeschlagenen Synthesewegen von TP3T werden durch die Literatur bestätigt. Dieses Ergebnis wurde in Nature Communications veröffentlicht.

Dieses Ergebnis wurde in Nature Communications veröffentlicht.
Link zum Artikel:
https://www.nature.com/articles/s41467-023-41698-5
Folgen Sie dem offiziellen Konto und antworten Sie mit „Retrosynthese“, um das vollständige Dokument als PDF zu erhalten
Experimentelle Verfahren
Algorithmuskonstruktion:Modul + Unterraster
Der gesamte retrosynthetische Analyseprozess umfasst vier Schritte: molekulare Grafikkodierung, Multitasking-Lernen, Entscheidungsfindung und Vorhersage der mehrstufigen Syntheseroute.
RetroExplainer besteht hauptsächlich aus vier Modulen: Multi-sensory Multi-scale Graph Transformer (MSMS-GT), Dynamic Adaptive Multi-task Learning (DAMT), Explainable Decision Module und Route Prediction Module.
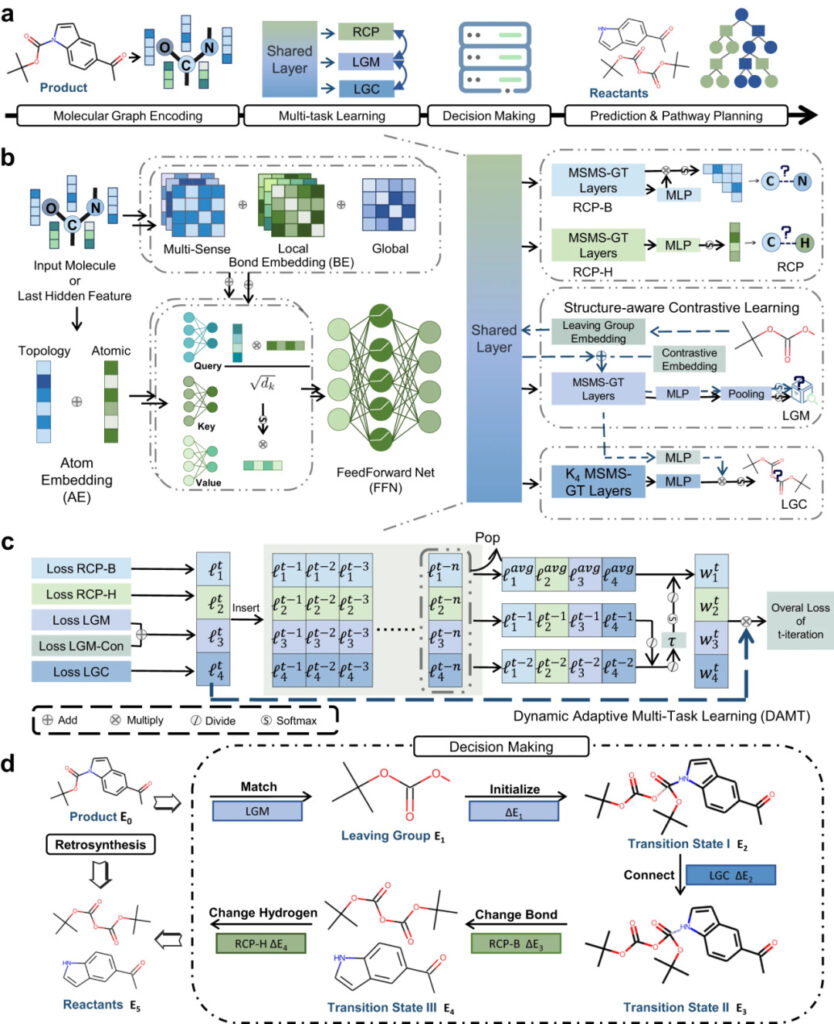
Abbildung 3: Schematische Darstellung von RetroExplainer und seinen Modulen
a: RetroExplainer-Prozessdiagramm;
b: MSMS-GT-Architektur;
c: Schematische Darstellung des DAMT-Algorithmus;
d: Entscheidungsprozess ähnlich dem Reaktionsmechanismus.
MSMS-GT erfasst wichtige chemische Informationen durch die Einbettung chemischer Bindungen und topologischer Einbettung von Atomen. Die kodierten Informationen werden durch den Multi-Head Attention-Mechanismus zu molekularen Vektoren verschmolzen.
Im DAMT-Modul werden molekulare Informationen gleichzeitig in die Untergitter Reaction Center Prediction (RCP), Leaving Group Match (LGM) und Leaving Group Connect (LGC) eingegeben.
RCP identifiziert Änderungen in chemischen Bindungen und die Anzahl der Wasserstoffatome neben Atomen, LGM gleicht die Abgangsgruppe im Produkt mit der in der Datenbank ab und LGC verknüpft die Abgangsgruppe mit dem Produktrest.
Das Entscheidungsmodul wandelt Produkte basierend auf den fünf retrosynthetischen Aktionen und dem Energiewert (E) der Entscheidungskurve in Reaktanten um und simuliert umgekehrt den molekularen Montageprozess.
Schließlich wird ein heuristischer Baumsuchalgorithmus verwendet, um einen effizienten Produktsyntheseweg zu finden und gleichzeitig die Verfügbarkeit der Reaktanten sicherzustellen.
Leistungsvergleich:USPTO-Benchmark-Datensatz
Um die Leistung von RetroExplainer zu überprüfen, verglichen die Forscher ihn mit 21 anderen Retrosynthese-Algorithmen auf Basis chemischer Reaktionen, die beim United States Patent and Trademark Office (USPTO) gelistet sind. Der Bewertungsindikator war dabei die Top-k-Genauigkeit.
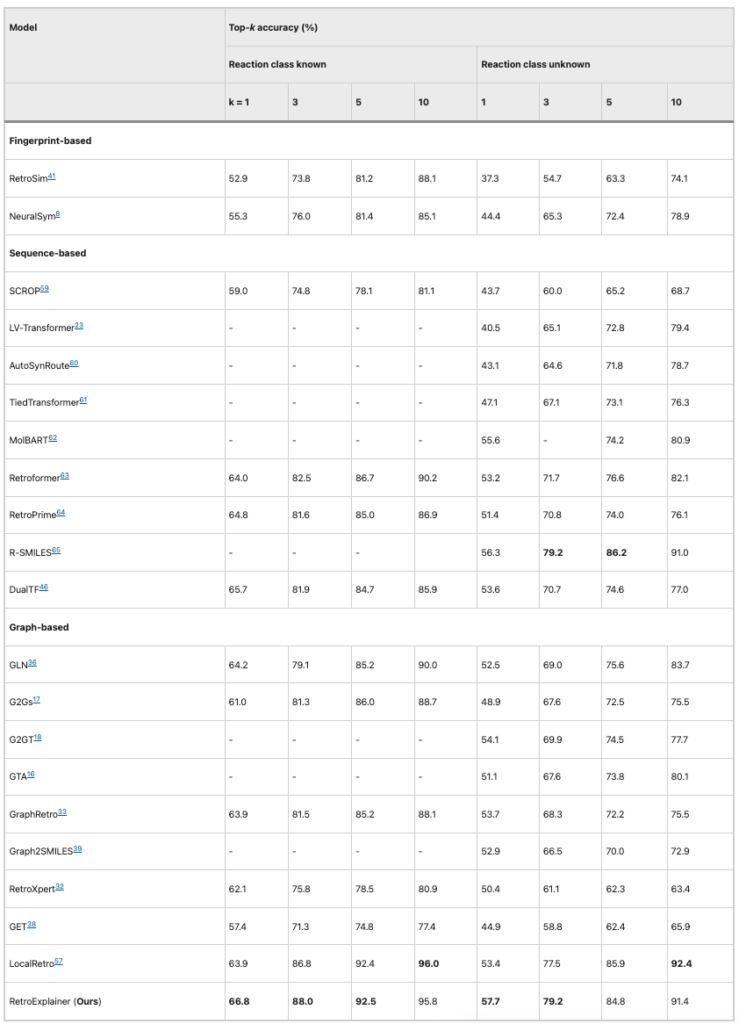
Tabelle 1: Leistungsvergleich von RetroExplainer und anderen Algorithmen (USPTO-50K)
Es ist ersichtlich, dass basierend auf dem USPTO-50K-Datensatz unter den 8 BewertungsindikatorenRetroExplainer übertrifft andere Algorithmen in 5 Aspekten und belegt den ersten Platz bei der durchschnittlichen Genauigkeit. Obwohl RetroExplainer in der Top-10-Genauigkeit LocalRetro unterlegen ist, beträgt der Abstand zwischen den beiden nur 1%.
Um den Einfluss ähnlicher Moleküle auszuschließen, haben die Forscher die Daten mithilfe von Tanimoto Similarity neu partitioniert und mit den beiden genauesten Algorithmen, R-SMILE und LocalRetro, verglichen.
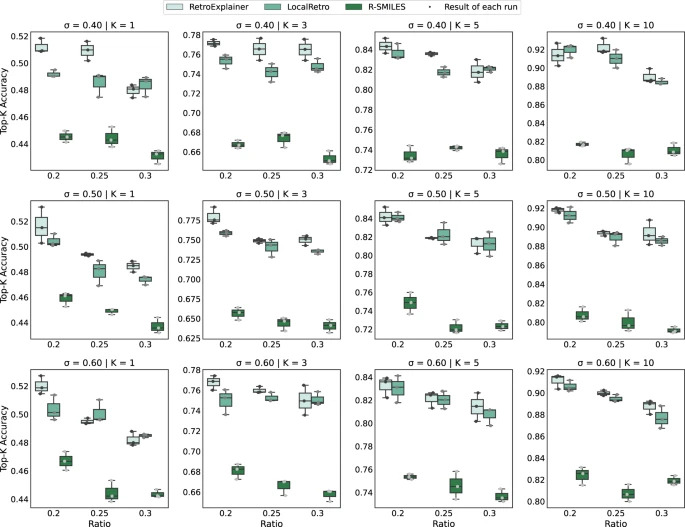
Abbildung 4: Leistungsvergleich von RetroExplainer, R-SMILES und LocalRetro anhand verschiedener Datensätze
Wie aus den Ergebnissen hervorgeht, schneidet RetroExplainer in den meisten Datensätzen besser ab, was seine Stabilität und Anpassungsfähigkeit widerspiegelt.
Anschließend verglichen die Forscher die Leistung des Algorithmus anhand der größeren Datensätze USPTO-MIT und USPTO-FULL. RetroExplainer übertrifft andere Algorithmen in allen Indikatoren, und der Abstand zu anderen Algorithmen ist sogar noch größer.Dies zeigt, dass RetroExplainer ein größeres Potenzial bei der Analyse umfangreicher Daten hat.
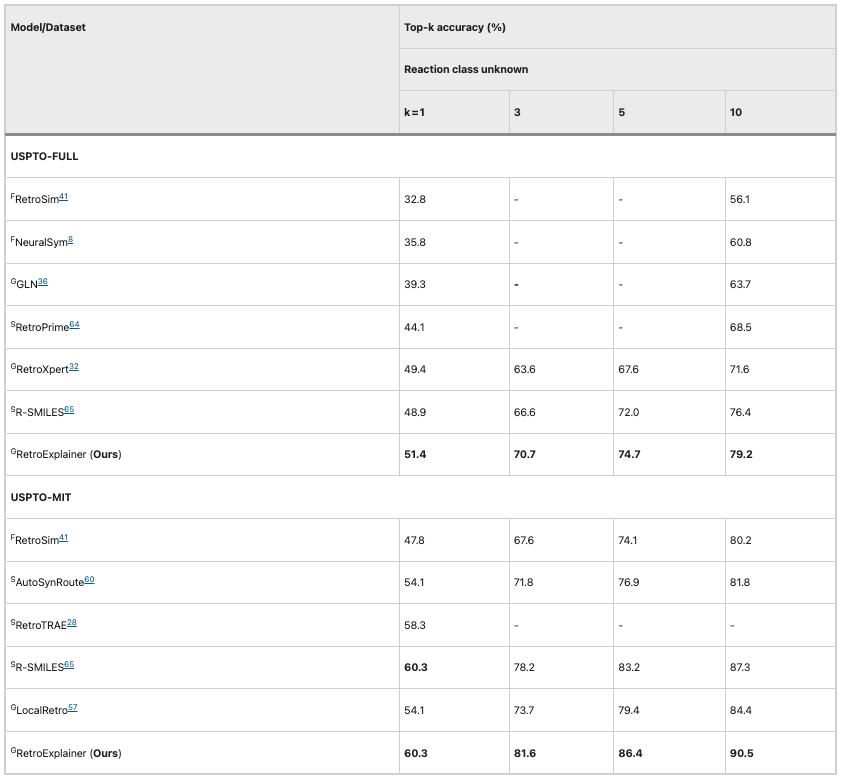
Tabelle 2: Leistungsvergleich von RetroExplainer und anderen Algorithmen (USPTO-MIT und USPTO-FULL)
Erklärbarkeit:Entscheidungsvisualisierung
Inspiriert von der bimolekularen nukleophilen Substitutionsreaktion (SN2) entwickelten die Forscher einen interpretierbaren retrosynthetischen Vorhersageprozess, der auf einer durch Deep Learning gesteuerten Molekülassemblierung basiert. Der Entscheidungsprozess umfasst sechs Phasen: Originalprodukt (P), Abgangsgruppen-Matching (S-LGM), Initialisierung (IT), Abgangsgruppen-Verbindung (S-LGC), Änderung der chemischen Bindung des Reaktionszentrums (S-RCP) und Änderung der Wasserstoffatomzahl (HC).
Basierend auf dem Beitrag jeder Phase zur endgültigen Entscheidung generiert das DAMT-Subgrid für jede Phase einen Energiewert (E).
Der konkrete Ablauf ist wie folgt:
1. In der P-Phase wird E jeder Phase auf 0 initialisiert;
2. In der S-LGM-Phase wird die Abgangsgruppe basierend auf der vorhergesagten Wahrscheinlichkeit des LGM-Moduls ausgewählt.
3. Addieren Sie das E der in der S-LGM-Phase ausgewählten Abgangsgruppe zur von den RCP- und LGM-Modulen vorhergesagten Antwortereigniswahrscheinlichkeit, um die Energie der IT-Phase zu erhalten.
4. In den Phasen S-LGC und S-RCP werden alle möglichen Knoten im Suchbaum basierend auf dem Algorithmus der dynamischen Programmierung erweitert. Wählen Sie Ereignisse mit einer Wahrscheinlichkeit größer als ein voreingestellter Schwellenwert aus und behalten Sie E bei;
5. Passen Sie die Anzahl der Wasserstoffatome und die formale Ladung jedes Atoms an, um sicherzustellen, dass das resultierende Moleküldiagramm den Valenzregeln entspricht, und berechnen Sie das endgültige E.
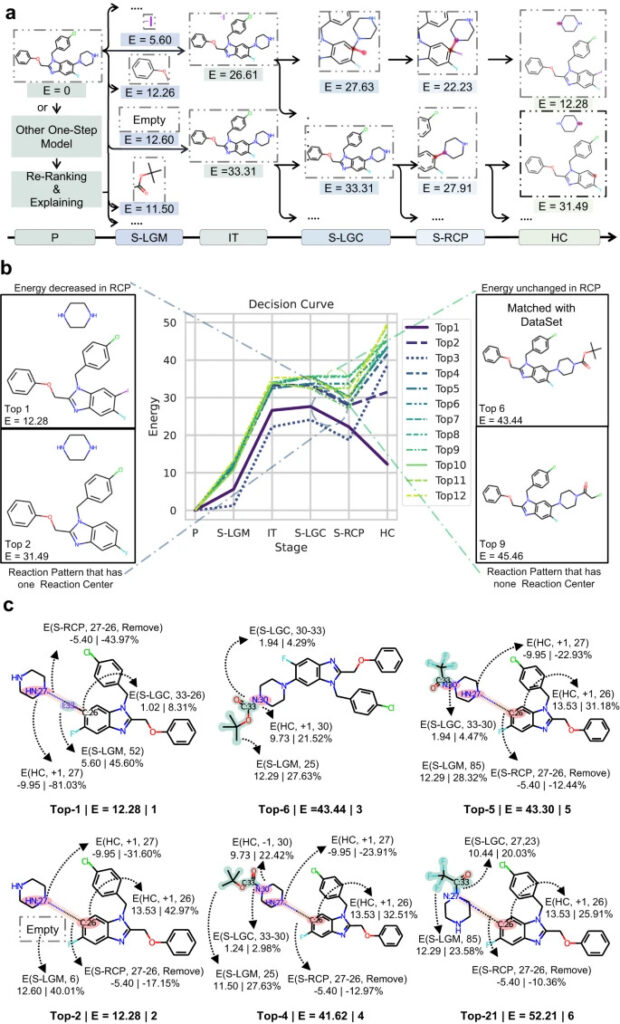
Abbildung 5: Der Entscheidungsprozess von RetroExplainer
a: Suchpfad von RetroExplainer für zwei Vorhersageergebnisse;
b: Entscheidungskurven der 12 besten vorhergesagten Routen;
c: 6 Strukturänderungsprozesse, die die Synthesewege darstellen.
Indem wir eine Entscheidungskurve basierend auf der Änderung von E zeichnen, können wir den Entscheidungsprozess von RetroExplainer analysieren und die Vorhersagefehler von RetroExplainer herausfinden.
Wie in der Abbildung gezeigt, sollte der richtige synthetische Weg zum Produkt die Entschützungsreaktion des Amins sein, RetroExplainer stuft sie jedoch an 6. Stelle ein, und die 1. ist die CN-Kopplungsreaktion. Die Analyse ergab, dass RetroExplainer dazu neigte, die Anzahl der Wasserstoffatome von Aminen im HC-Stadium zu erhöhen, was zu diesem Unterschied führte.Dies deutet darauf hin, dass RetroExplainer im HC-Stadium möglicherweise die gleiche Fehleinschätzung bei Molekülen mit ähnlicher Struktur vornimmt.
Durch den Vergleich der Reaktionen von RetroExplainer auf Platz 1 und 2,Forscher fanden heraus, dass E mit der Schwierigkeit der Antwort zusammenhängen könnte. Obwohl die Verbindung von I:33 und C:26 in Reaktion 1 nicht zur Energiereduzierung beiträgt, erfordert die Verbindung eines Wasserstoffatoms an C:26 die 13-fache Energie der vorherigen Reaktion. Gleichzeitig schwächte die Einführung von I:33 das Selektivitätsproblem der CN-Kupplungsreaktion ab.
gleichzeitig,Sterische Hinderung kann auch die Vorhersageergebnisse von RetroExplainer beeinflussen. Vergleicht man die Reaktionen auf Platz 4 und Platz 21, so sind ihre Molekülstrukturen gleich, aber die Abgangsgruppen sind an das symmetrische N gebunden, was zu Unterschieden in E führt.
Pfadplanung:Mehrstufige vorhergesagte Synthesewege
Um die Praktikabilität der Vorhersagen von RetroExplainer zu verbessern, kombinierten die Forscher es mit dem Retro-Algorithmus und ersetzten die einstufigen Vorhersagen des letzteren durch mehrstufige Vorhersagen.
Am Beispiel des Bronchodilatators Protokylol hat RetroExplainer einen vierstufigen Syntheseweg für dieses Produkt entwickelt. Anschließend führten die Forscher eine Literaturrecherche zu diesen vierstufigen Reaktionen durch, um ihre Durchführbarkeit zu untersuchen.
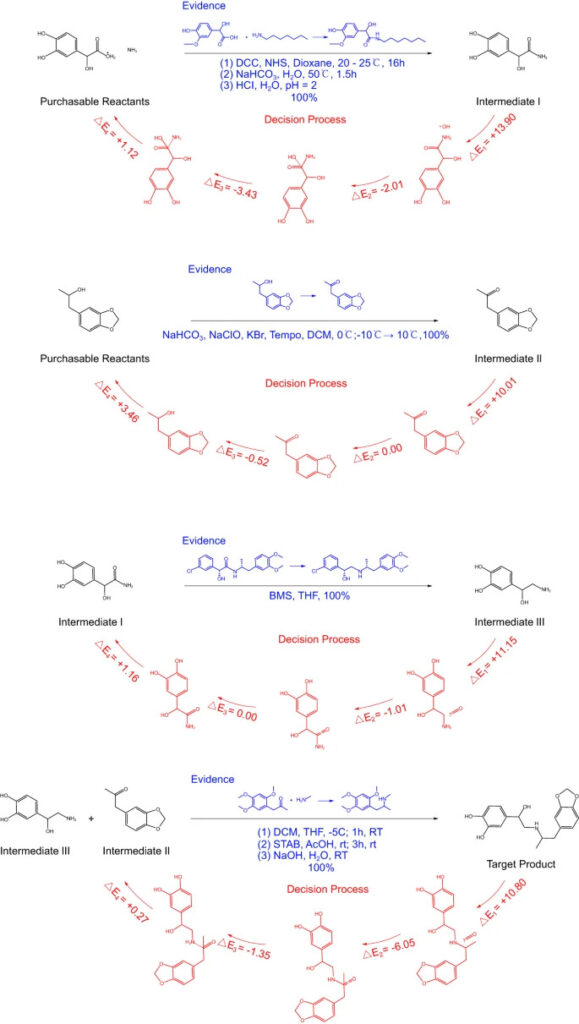
Abbildung 6: RetroExplainers 4-stufige Synthese von Protosan
Der blaue Text in der Abbildung ist eine ähnliche Antwort, die in der Referenz aufgezeichnet wurde, und der rote Teil ist der Entscheidungsprozess von RetroExplainer.
Obwohl bei vielen Reaktionen nicht genau die gleiche Referenz gefunden wurde, ergaben sich ähnliche Reaktionen mit hoher Ausbeute. Auch,RetroExplainer hat 176 Experimente für 101 Fälle entworfen, von denen 153 ähnliche Reaktionen in SciFinder finden konnten.
Die obigen Ergebnisse zeigen, dass die Retrosynthesevorhersage von RetroExplainer besser ist als bei anderen aktuellen Algorithmen. Gleichzeitig sind die Entscheidungen von RetroExplainer transparent und erklärbar und es führt eine mehrstufige Planung der Antworten durch, wodurch diese praktikabler wird. RetroExplainer soll ein leistungsfähiges Werkzeug für die retrosynthetische Forschung in der organischen Chemie darstellen.
Leistung vs. Erklärbarkeit, der Widerspruch der KI
Erklärbarkeit ist ein Schlüsselfaktor bei der Anwendung von KI in verschiedenen Szenarien. Mit der kontinuierlichen Entwicklung der KI in Branchen wie autonomes Fahren, medizinische Diagnose, Finanzen und Versicherungen hat der Entscheidungsprozess der KI zunehmend an Bedeutung gewonnen und ist mit immer mehr praktischen, sozialen und sogar rechtlichen Problemen konfrontiert.
Gleichzeitig kann die Erklärbarkeit den Benutzern helfen, KI zu verstehen, zu warten und zu verwenden sowie neue Konzepte in KI-Anwendungsfeldern zu entdecken und zu verstehen. Durch die Erklärbarkeit wird außerdem die Umsetzbarkeit der Ergebnisse aufgezeigt und den Benutzern mitgeteilt, welche Entscheidung den größten Nutzen bringt.
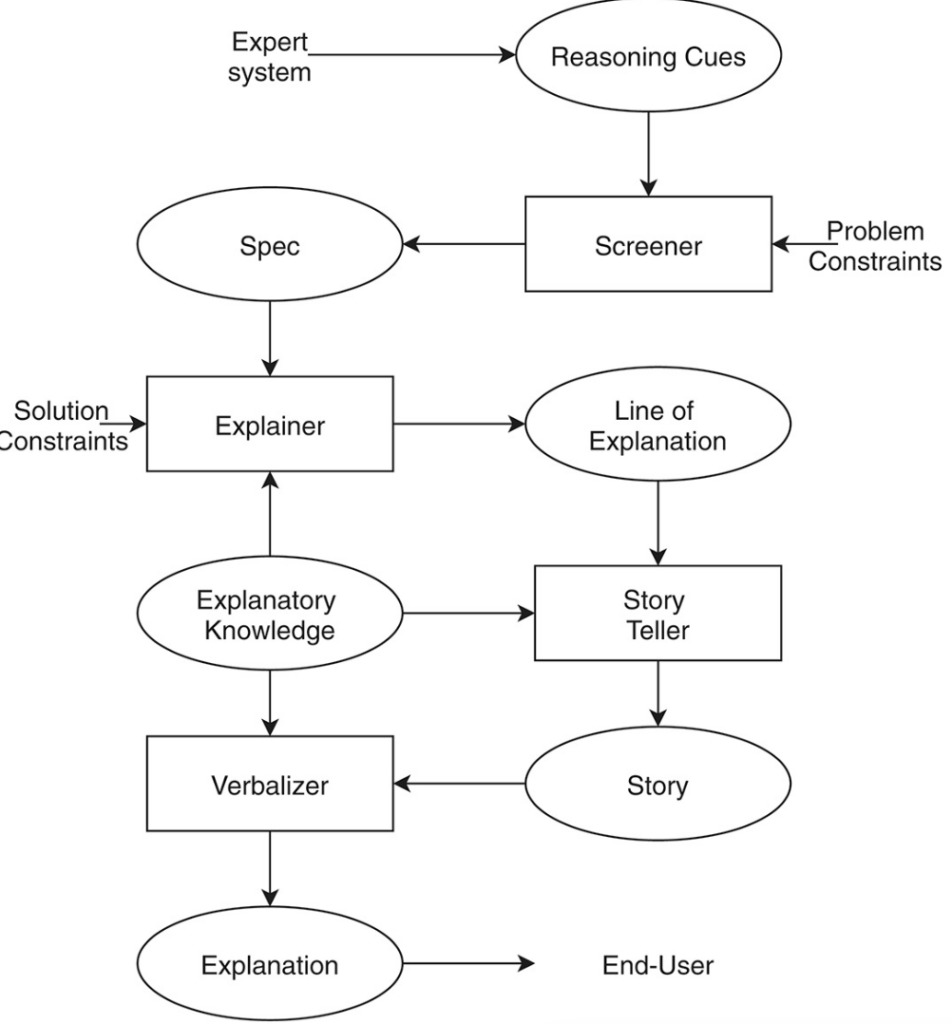
Abbildung 7: Erklärungsphase des Problemlösungsprozesses
Jedoch,Modellleistung und Modellinterpretierbarkeit sind ein großes Problem für ScienceAIWenn das Modell eine gute Leistung und eine gute Robustheit gegenüber den Testsätzen aufweist, funktionieren hochdimensionale Deep Features möglicherweise besser, haben jedoch keine physikalische Bedeutung, was wir oft als "Die Interpretierbarkeit wissenschaftlicher Forschung ist im Allgemeinen schlecht".
Im Gegenteil: Wenn gut erklärte Merkmale verwendet werden, ist die tatsächliche Modellleistung zwar physikalisch sehr gut interpretierbar, hängt jedoch stark von den Daten ab und nimmt bei einer Änderung des Datensatzes ab.
Es gibt noch keine gute Möglichkeit, den Widerspruch zwischen den beiden zu überbrücken, aber in dieser Studie haben die Forscher den Entscheidungsprozess der KI Schritt für Schritt visualisiert., sodass Benutzer die Punkteänderungen verschiedener Vorhersageergebnisse in jeder Phase klar nachvollziehen können, den Entscheidungsprozess der KI verstehen und Entwicklern die Optimierung des Modells erleichtern können.
Mit der kontinuierlichen Entwicklung erklärbarer KI wird das Verständnis der Menschen für KI tiefer und der Entscheidungsprozess von KI wird leichter verständlich.In Zukunft wird die Interaktion zwischen Mensch und Maschine weiter zunehmen, die Hemmschwelle zur Interaktion wird weiter sinken und KI wird in mehr Szenarien eingesetzt, wodurch das Leben bequemer und intelligenter wird.
Referenzlinks:
[1]http://www.chem.ucla.edu/~harding/IGOC/R/retrosynthesis.html
[2] https://zh.wikipedia.org/zh-cn/Vereinfachte molekulare lineare Eingabespezifikation
[3]https://wires.onlinelibrary.wiley.com/doi/10.1002/widm.1391








