Command Palette
Search for a command to run...
計算コストが半分に削減された化学反応発見ツール ChemOntology は、人間の直感をシステムに「エンコード」し、反応経路の探索を加速します。
化学反応機構は、物質変換を支配する本質的な法則を明らかにするだけでなく、効率的な触媒の設計やグリーン合成経路の開発といった産業応用にとって重要な証拠を提供します。反応機構の解析は、重要な計算手法である反応経路探索に大きく依存しています。これは、ポテンシャルエネルギー面(PES)上で局所的最小値と反応中間体を特定し、実際の反応経路をマッピングするものです。
計算化学者は長年にわたり、有限配置を生成することで反応機構を探索するために、主に固有反応座標(IRC)法に頼ってきました。しかし、この従来のアプローチには重大な限界があります。研究者が事前に設定した経路に制約されることが多く、非従来的な反応経路を見落としやすく、結果として潜在的な代替機構を見逃す可能性があります。
人工力誘起応答(AFIR)などの自動化手法の開発により、バイアスのない応答経路探索が可能になりました。これらの手法は、応答経路を相互接続された「ノード」のネットワークとして扱い、新しい構成を反復的に生成することで応答の可能性を体系的に探索し、未知の応答メカニズムを発見するための新たな窓を開きます。
しかし、自動経路探索は完璧な解決策ではありません。多数の構成に対するエネルギー計算はコストが高く、さらに構造変化の背後にあるメカニズムを研究する必要があるため、計算負荷はさらに増大します。半経験的手法や機械学習によるポテンシャル関数はコストを部分的に削減できますが、エネルギー予測における時折の不正確さは、経路探索の信頼性に影響を与える可能性があります。
化学オントロジーは、「知識構造化ツール」として、前述のボトルネックを克服するための新たなアプローチを提供します。エンティティ、属性、関係性、ルールの標準化された定義を通じて、断片化された化学知識を機械可読かつ処理可能な構造化情報へと整理します。例えば、RXNOなどのオントロジーフレームワークは、反応経路のアノテーションにおいて既にその価値を実証しています。
これを基に、日本の北海道大学の研究チームが新しい AI システム「ChemOntology」を開発しました。化学知識分類システムとして、人間の化学的推論を機械が理解できるフレームワークに形式化し、それによって化学反応の迅速な調査と分析を可能にします。古典的なヘック反応機構の研究におけるこのフレームワークの適用の成功は、経路探索の加速におけるその有効性を検証するだけでなく、「人間の化学知識」と「自動計算」を統合する大きな可能性を浮き彫りにします。
「ChemOntology: 反応経路検索を迅速化する再利用可能な明示的化学オントロジーベースの方法」と題された関連研究結果が ACS Catalysis に掲載されました。
研究のハイライト
* トレーニング データセットに依存せずに、人間の化学者の直感をシステムに「プログラム」することに成功しました。これは、従来の機械学習手法に比べて大きな利点です。
* 実験結果によると、AFIR と組み合わせると、ChemOntology は約半分の数のパスを探索するときに完全な AFIR_TARGET 検索に匹敵する結果を達成でき、全体的な計算コストをほぼ半分に削減できます。
用紙のアドレス:
https://pubs.acs.org/doi/10.1021/acscatal.5c06298
完全な PDF を取得するには、当社の公式 WeChat アカウントをフォローし、バックグラウンドで「ChemOntology」と返信してください。
AIフロンティアに関するその他の論文:
https://hyper.ai/papers
知識駆動型フレームワークのデータ方法論
この研究機関が利用するデータリソースは、機械学習モデルの学習に従来用いられてきた大規模なデータセットではありません。これはまさに、知識駆動型フレームワークであるChemOntologyの固有の特性によるものです。ChemOntologyは、データのフィッティングに依存せず、化学的なルールとメカニズムに焦点を当てているため、大規模データへの依存度が高く、方法論レベルでの潜在的な限界を回避できます。
初め、研究者らは、公開化学データベース PubChem を使用して、反応におけるすべての主要成分に関する標準化された情報を入手しました。これには、分子構造、名称、そして固有の識別子が含まれます。これらの情報は、各化学物質の「IDカード」とも言えます。反応系における各成分の役割を正確に定義するのに役立つだけでなく、固有の化合物番号によって標的生成物を追跡し、無関係または不要な副生成物を除去することも可能になり、後続の反応経路の探索をより正確かつ効率的に行うことができます。
第二に、実際の複雑な化学シナリオにおけるこの方法の信頼性と適用性をテストするために、研究者らは、多様なメカニズムと多数の反応ステップを持つ古典的なヘック反応をテストケースとして選択した。システムには、反応物、触媒、配位子、塩基の3次元構造ファイル、既知の中間体および最終生成物の参照エネルギーデータなど、完全な入力情報が提供されました。この代表的な事例では、複雑な反応ネットワークにおける本手法の性能を徹底的に検証し、主要な中間体の同定能力や主反応経路と副反応経路の区別を検証するだけでなく、計算コストの削減における利点を直感的に示しています。
全体として、本研究では、信頼できるデータベースを通じて情報の正確性を確保し、典型的な複雑な反応を活用して方法の有効性を検証し、完全なオープンソースを通じてコラボレーションと反復を促進し、大規模なトレーニングデータに依存せずにさまざまな有機金属反応システムへの幅広い適用性を維持できるようにしています。
ChemOntology: 有機金属反応における経路探索のための新しいフレームワーク
ChemOntology は、大規模なデータによるモデルのトレーニングに依存しない、知識駆動型コンピューティング フレームワークです。代わりに、化学反応ルール、構造的制約、量子化学経路探索プロセスを体系的に統合します。これにより、定義された化学的コンテキスト内での反応経路の効率的な探索が可能になります。この手法は、計算エンジンとしてAFIR(人工力誘起反応)を使用し、化学的知識を明示的にエンコードして探索方向を導き、生成された構造をリアルタイムでスクリーニングすることで、無意味または不合理な反応展開を回避します。
下の図に示すように、ChemOntology ワークフローは、ユーザー入力の解析、セットアップ ファイルでの化学情報のモデリング、ERPO を使用した反応パスの生成、構造合理性の制約、AFIR の実行と制御、およびパス分析で構成されています。
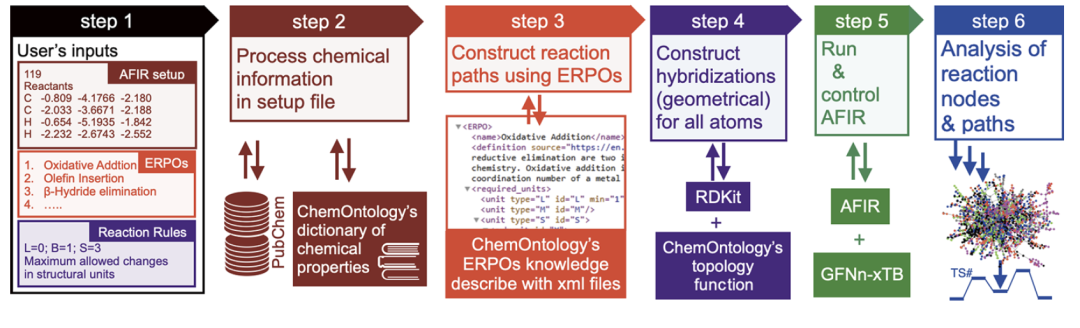
反応システムはまず、金属、配位子、基質、および任意の塩基などの構造単位の集合として分析され、各タイプの単位には特定の化学的役割と特性が割り当てられます。反応プロセスは、構造単位とその内部原子の混成状態の漸進的な変化として記述され、「反応ノード - 構造単位 - 原子」という3つのレベルで構造変化を追跡します。この階層的表現により、モデルは電子構造の詳細に依存せず、幾何学的および位相的情報のみに基づいて反応経路の化学的妥当性を判断することができます。
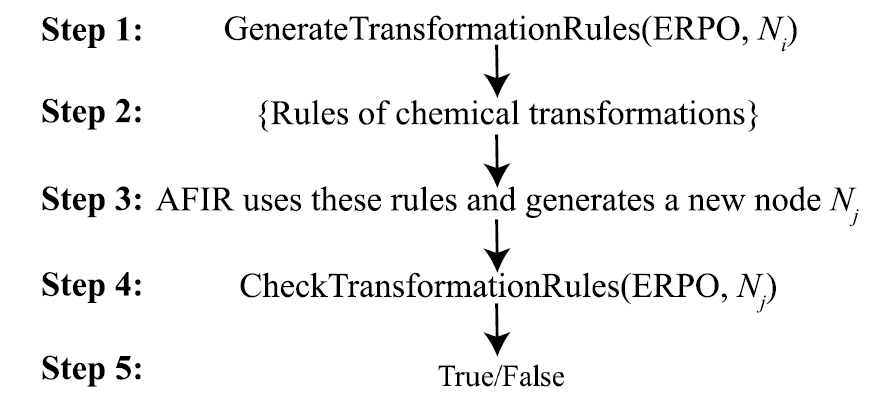
反応経路の生成は、ERPO (Elementary Reaction Pathway Operator) に依存します。つまり、一般的な有機金属の基本反応プロセスのモジュール記述です。反応の例としては、配位化合物の形成、酸化付加、オレフィン挿入、β水素脱離などが挙げられます。ERPOは反応配列の構築だけでなく、検索プロセスにおけるルール検証ツールとしても機能し、各構造変化が期待される化学的意味論に適合していることを確認します。複雑な反応をコンビナトリアルな基本プロセスに分解することで、ChemOntologyは反応の多様性を維持しながら、検索空間におけるコンビナトリアルな複雑さを大幅に低減できます。
反応の進化をさらに制限するために、ChemOntology は、原子混成の変化に基づいたフィルタリング メカニズムを導入します。ユーザーは、いくつかのパラメータを設定することで、反応プロセス全体を通して、様々な構造単位の最大許容構造調整量を制限できます。制約を超える幾何構造は自動的に識別され、探索から除外されます。このメカニズムにより、構造爆発の問題が効果的に抑制され、特定の反応結果を事前に設定することなく計算効率が大幅に向上します。
実際の計算では、ChemOntologyはAFIR探索プロセスの上位に知識制御層として組み込まれ、半経験的タイトバインディング法GFN2-xTBと組み合わせて反応経路の幾何学的進化を記述します。機械学習モデルとは異なり、ChemOntology ではデータセットによるトレーニングは必要ありません。その「知識ベース」は主に、官能基認識ルール、構造単位分類スキーム、および ERPO ファイルで構成されています。これらはすべて、研究対象に応じてユーザーが柔軟に変更できます。この設計により、ChemOntologyは計算化学の方法論に近いものとなり、自動化された反応探索プロセスに人間の化学的直感を体系的に導入するために使用されます。
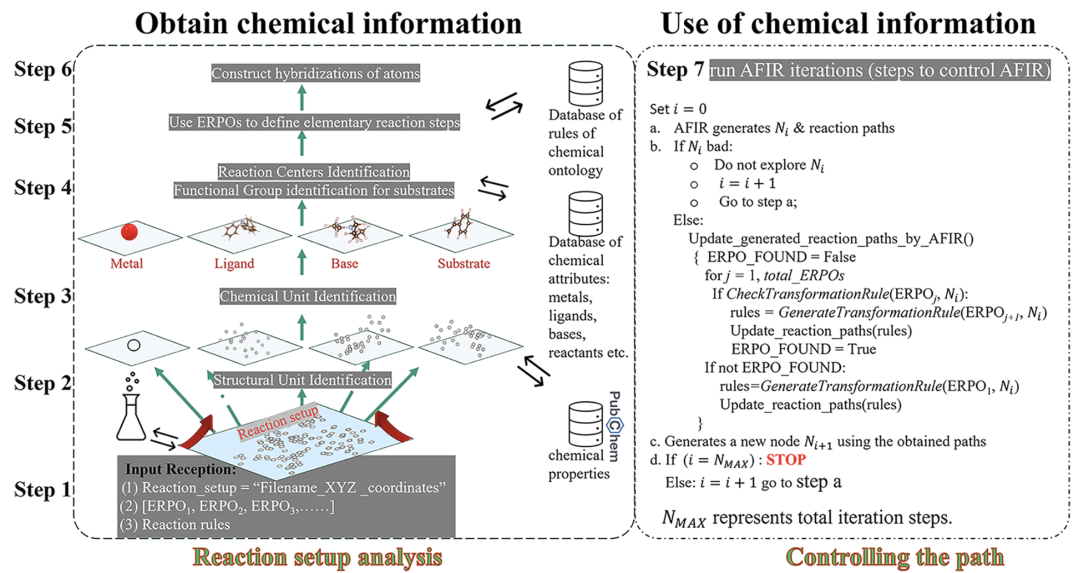
全体として、ChemOntology は、明示的な化学的制約の下で反応経路を検索するためのプラットフォームを提供します。新しい反応性の出現を制限するのではなく、構造化されたルールを通じて「合理的な化学空間」内で探索するように計算をガイドし、それによって反応メカニズムの分析と潜在的な新しい化学物質の発見のバランスを実現します。
実験結果: 計算コストが半分になり、パスの明瞭度が 2 倍になりました。
反応経路探索におけるChemOntologyフレームワークの有効性と効率性を検証するために、研究チームは、複雑なメカニズムを持ち、代表的な古典的なヘック反応を試験系として選択した。下図に示すように、この反応はヨードベンゼンとスチレンを基質として用います。パラジウム触媒、トリフェニルホスフィン配位子、トリエチルアミン塩基の条件下では、主にトランススチルベンが生成され、少量のシス異性体と微量の副生成物が伴います。その反応機構は、酸化付加、オレフィン挿入、移動挿入、β水素脱離、塩基脱離など、複数の重要なステップから構成されています。反応中心の数が多すぎるため、自動化経路探索法にとって典型的な課題となっています。
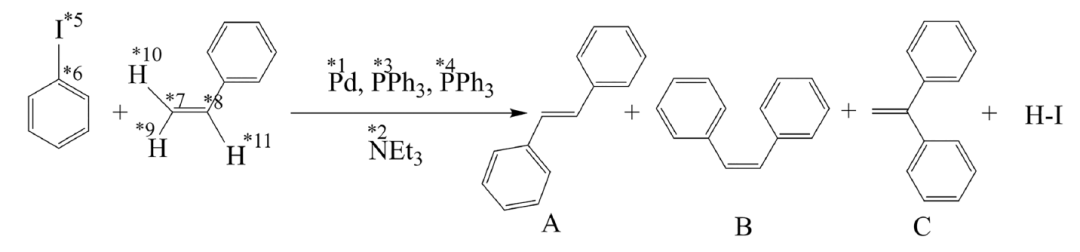
本研究では、3つの並列パス探索戦略、すなわち、誘導なしのAFIR_DEFAULT、部分的に制約のあるAFIR_TARGET、そして化学オントロジーを組み込んだAFIR_ChemOntologyを比較しました。これら3つの戦略は、「知性」のレベルにおいて根本的に異なります。前者は構成空間をほぼ無差別に探索するのに対し、後者は人為的な制約によって探索範囲を絞り込みます。一方、AFIR_ChemOntology は、そのフレームワークを通じて反応物と主要な反応中心の化学的役割を自動的に識別し、基本的な反応プロセスを活用して検索を動的にガイドします。
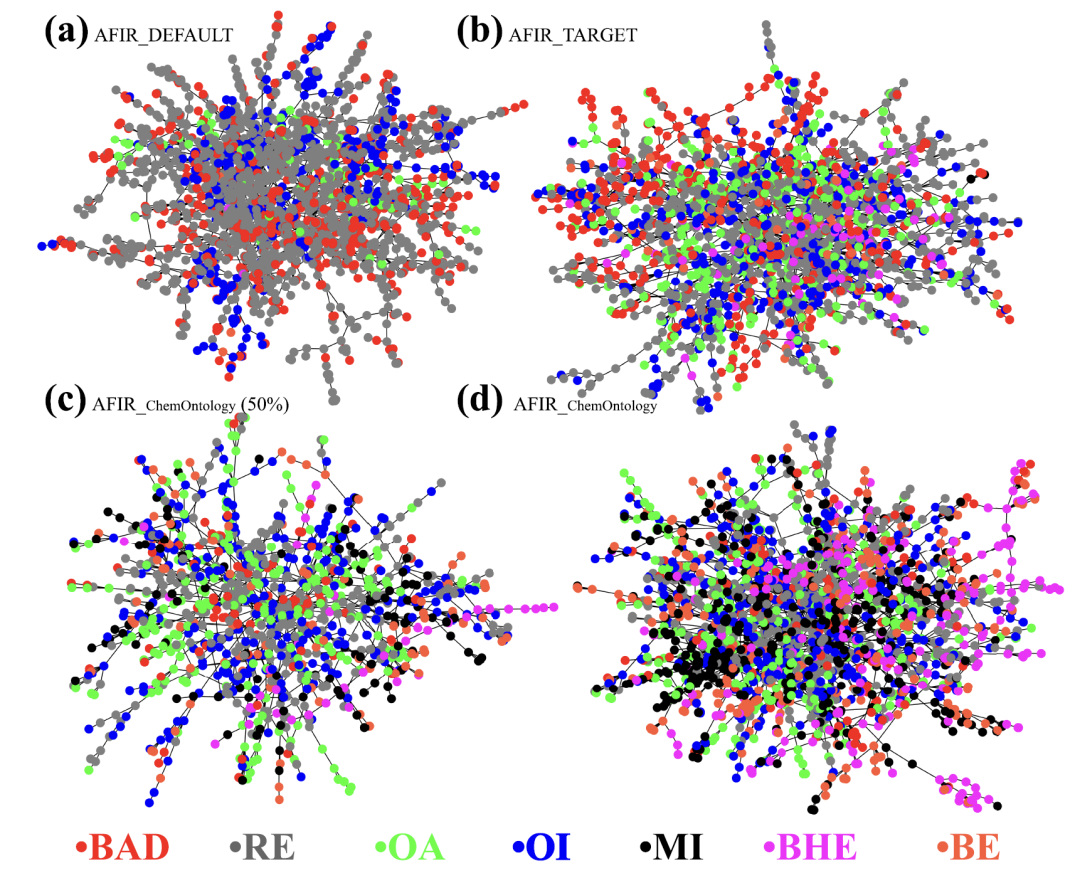
下図に示すように、同じ計算条件下において、3つの手法で生成された反応ネットワークは大きく異なります。AFIR_DEFAULTは化学的に意味のない無効なノードを大量に生成し、有効な経路を著しく飽和させています。AFIR_TARGETはいくらか改善が見られますが、依然として多くの冗長な構造が残っています。対照的に、AFIR_ChemOntology の検索結果は非常に焦点が絞られており、主要な反応経路を早期に明確に特定できます。計算は化学的に妥当な経路に焦点を当てて行われました。さらに中間体統計を分析したところ、ChemOntologyによって「不良ノード」の割合が大幅に減少し、特定された主要な中間体はHeck反応の古典的なメカニズムと非常によく一致していることが示されました。
下の図に示すように、エネルギー解析により、3 つの方法すべてが反応の初期段階における共通のステップを捉えていることがわかります。ただし、主生成物と副生成物に至る特定の経路をそれぞれ完全に区別して追跡できるのは AFIR_ChemOntology だけです。さらに、β水素除去に関連する特徴的な相互作用は、効率的な経路で一般的に観察されましたが、微量生成物につながる経路では、これらの相互作用はより弱い構造安定性を示し、それがそれらの生成確率が低いことを説明できるかもしれません。
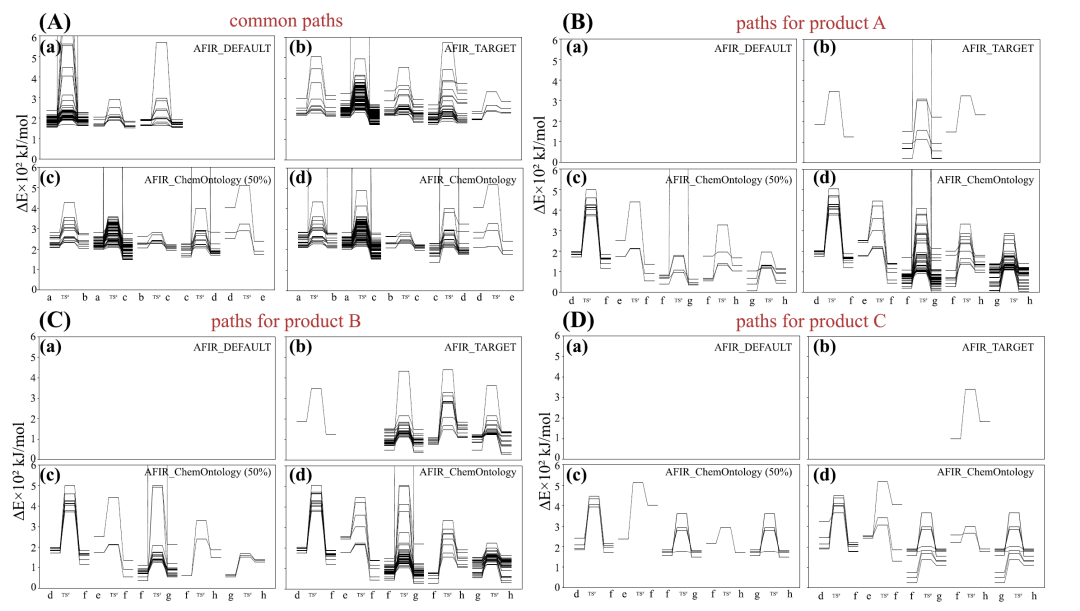
計算効率の観点から言えば、AFIR_ChemOntology は、AFIR_TARGET の完全検索と同等の効率を実現しながら、探索するパスの数を約半分に抑え、全体的な計算コストをほぼ半分に削減します。この利点は、主に化学知識による探索方向のガイダンスと、無効な構造のリアルタイムフィルタリングに由来します。全体として、実験結果は、化学オントロジーを自動経路探索に統合することで、化学的合理性を確保しながら機構解析の効率を大幅に向上させ、複雑な反応系の研究においてより効率的で信頼性の高いアプローチを提供できることを示しています。
研究室から工場へ:化学オントロジーによる反応探索の道筋の再構築
化学オントロジーと反応経路の自動検索の統合は、理論化学と産業応用を繋ぐ重要な架け橋を築きつつあります。この傾向は、学術界における一連の最先端研究を促しただけでなく、産業界においても重要な革新的実践を促し、反応機構研究を従来の「事後分析」からより積極的な「能動的なガイダンス」へと変革させています。
学術界では、アルゴリズムの革新とメカニズムの改良に研究が重点的に行われ、この分野の知識の限界を継続的に拡大しています。例えば、アイスランド大学のチームは「最適輸送ガウス過程」(OT-GP)アルゴリズムを開発しました。その中核は、一定量のトレーニング データのみを使用して効率的に機能できるインテリジェントなデータ フィルタリング戦略を採用することにあります。このアルゴリズムにより、分子反応経路探索の平均時間が 28.3 分から 12.6 分に大幅に短縮され、成功率も大幅に向上し、複雑なシステムの迅速なメカニズム探索のための新しいツールが提供されます。
論文タイトル: ガウス過程加速鞍点探索における堅牢性の向上と計算オーバーヘッドの削減のための適応的枝刈り
ペーパーリンク:https://doi.org/10.48550/arXiv.2510.06030
同時に、スイスのETHチューリッヒの研究チームは、第一原理分子動力学と強化されたサンプリング方法を組み合わせました。我々は、分子ふるいと遷移金属の触媒反応における重要な水素移動と転位のステップを体系的に研究し、反応環境による反応チャネルの動的変化の機構特性を明らかにし、触媒の合理的設計を導くために使用できる一般的な微視的図を提案した。
論文タイトル: 不均一触媒における強化サンプリングを用いた第一原理分子動力学
論文リンク:https://pubs.rsc.org/en/content/articlelanding/2022/cy/d1cy01329g
一方、産業界の実践は、これらの理論を実際の生産性に転換することに重点を置いています。米国の計算化学分野を代表する企業であるシュレーディンガー社を例に挙げてみましょう。AutoRW 自動反応ワークフローは、化学オントロジーの構造化された思考を深く統合します。反応の列挙とパスのマッピングから結果の整理と出力まで、プロセス全体の自動化を実現します。
一方、ドイツの化学大手BASFとIBMの提携も同様の技術統合の道筋を示している。両者は化学オントロジーと量子化学計算、人工知能を組み合わせ、高性能触媒の研究開発に共同で取り組みます。「知識誘導+AIコンピューティング」モデルの採用により、研究開発サイクルが大幅に短縮され、実験の試行錯誤にかかるコストが削減されただけでなく、自動車、建設などの分野におけるポリウレタン材料の応用のための強固な基盤も築かれました。
世界をリードする企業によるこれらの実践は、化学オントロジーと自動コンピューティングを組み合わせることの普遍的な価値を実証するだけでなく、地域間および分野間の技術協力を通じて、学術的なブレークスルーから技術移転、そして産業アプリケーションと需要のフィードバックへの好循環を形成し、世界の化学産業をより環境に優しく、より効率的で、よりスマートな未来へと継続的に推進しています。
参考リンク:
1.https://wp-stg.schrodinger.com/wp-content/uploads/2023/10/A4-22_111-Reaction-Workflow-Application-Note_R3-1-1.pdf
2.https://blog.csdn.net/cainiao080605/article/details/147259567
3.https://phys.org/news/2025-12-ai-mimics-human-intuition-explore.html








