Command Palette
Search for a command to run...
ICML 2025に選ばれた清華大学/人民大学は、統合型バイオ分子動力学シミュレータであるUniSimを提案した。

清華大学の劉楊教授のグループと中国人民大学高陵人工知能学院の黄文兵教授のグループは共同で、統合型バイオ分子時間粗化ダイナミクスシミュレーター UniSim を提案しました。この方法は、大量の3D分子構造データに対するノイズ除去+力場ハイブリッド事前トレーニングを通じて統一された全原子表現モデルを取得し、確率的補間生成フレームワークに基づいて長い時間ステップで分子の移動ベクトル場を学習し、さまざまな化学環境に迅速に適応するための力誘導コアを導入します。 UniSim は、分子タイプ (小分子、ペプチド、タンパク質) と化学環境全体にわたって、統一された時間粗化ダイナミクス シミュレーション フレームワークを実現した最初の製品です。分子シミュレーション分野におけるディープラーニングの実用化を推進しました。
関連する成果は、「UniSim: 生体分子の時間粗大化ダイナミクスのための統合シミュレータ」というタイトルで ICML 2025 に選出されました。

用紙のアドレス:
AIフロンティアに関するその他の論文:
https://go.hyper.ai/UuE1o
統一された時間的粗大化シミュレータがなぜ必要なのでしょうか?
研究者たちは、分子動力学シミュレーションの分野では、統一された時間粗大化シミュレータを構築することが合理的かつ必要であると考えています。一方、統一されたモデリング フレームワークは、分子システム全体にわたる共同シミュレーションの基盤となります。例えば、タンパク質-リガンド相互作用のような複雑系のシミュレーションでは、タンパク質と低分子が同一の物理環境に共存することがよくあります。モデルが特定の種類の分子にしか適用できない場合、両者の相互作用挙動を全原子スケールで正確に再現することは困難です。そのため、統一的な表現機能を備えたシミュレータは、同一のモデルフレームワーク内で複数の種類の分子を同時に扱うことができ、多分子複合体のモデリングのための強固な基盤を提供します。
一方、統一モデルは、さまざまな種類の分子の構造データと動的データを統合するのに役立ち、それによってモデルの一般化と転送機能が向上します。現在利用可能な分子軌道データは非常に希少かつ不均一に分布しており、タンパク質、ペプチド、低分子など、様々な種類のデータがそれぞれ独自の強みを持っています。これら全てを同じモデルの事前学習と学習に活用できれば、モデルの原子レベルの構造理解が飛躍的に向上し、分子間ドメインマイグレーション能力も強化されます。
同時に、時間粗大化シミュレーションを導入することも、シミュレーション効率を向上させるための中核的な方法です。従来の分子動力学シミュレーションは、極めて小さな時間ステップ(フェムト秒など)で段階的に計算を進めるため、計算コストが高く、タンパク質の折り畳みなどの長期的な挙動をカバーすることが困難です。時間粗大化法は、現在の状態から将来の状態へのマッピング関係を直接学習します。物理的な一貫性を維持するという前提の下、従来のステップサイズよりもはるかに大きな時間スケールで迅速に軌跡を生成することができるため、シミュレーション効率が大幅に向上し、実用的な時間で長期的なシミュレーションを実行することが可能になります。
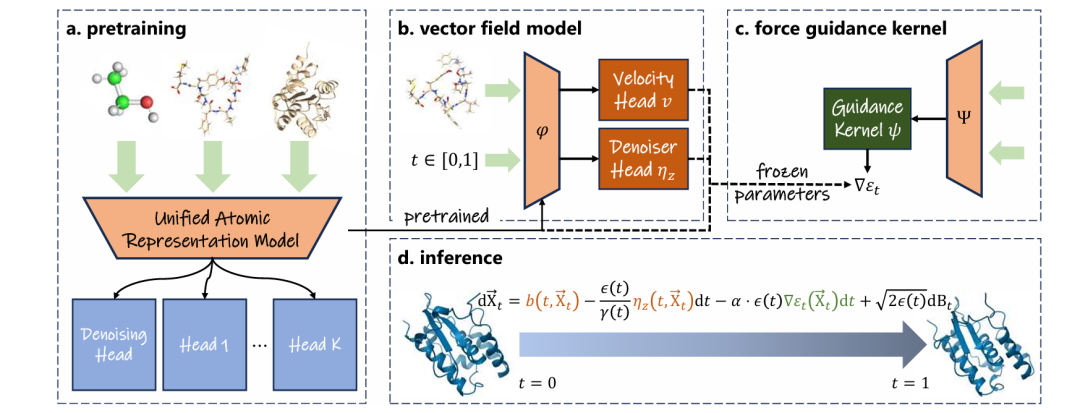
統一表現:マルチスケールおよびマルチタイプの分子の特性評価の問題を解決する
統一された全原子表現モデルは分子種間の動力学シミュレーションの基礎であるが、ただし、このようなモデルの実装には、依然として次のような技術的な課題が存在します。
* まず、分子システムは、数十個の原子からなる小さな有機分子から数千個の原子からなるタンパク質高分子まで多岐にわたり、規模が大きく異なり、構造が複雑かつ多様です。すべての原子を直接トレーニングに使用すると、モデルは分子の種類ごとに異なる注意メカニズムを持つことになり、モデルの相互転送能力が阻害されます。
* 第二に、統一された全原子表現を実現するための基礎は、統一された原子レベルの語彙を使用することです。直感的なアプローチとしては、周期表を埋め込み表現の語彙として直接用いることが挙げられます。しかし、このアプローチでは、天然アミノ酸などの部分構造など、ペプチドやタンパク質に多数存在する規則的な単位が無視されるため、タンパク質型データではパフォーマンスが低下します。
* 最後に、さまざまな状態の分子の表現を完全に学習するために、大量の定常状態および非定常状態の分子 3D 構造データが事前トレーニング データセットに含められます。不安定な分子を事前学習するための一般的なパラダイムは、原子に作用する力を学習することです。しかし、データセットによって原子力場を計算する際に異なる力場パラメータが使用され、ラベルデータの不整合が生じます。
統一されたモデリングを実現するために、UniSim は上記の問題を解決するための 3 つの主要なテクノロジを導入しています。
* 勾配環境サブグラフ: 分子スケールを適切にバランスさせる
データ前処理段階では、大きな分子(1,000 個以上の原子を含む)の 3D 構造データがセグメント化されます。分 < r最大 前処理中に、分子内の任意の原子がランダムに選択され、その原子が球の中心として扱われます。分 そしてr最大 半径の球体を作る小さなボールに含まれる原子は勾配サブグラフと見なされ、大きなボールに含まれる原子は環境サブグラフと見なされます。原子間の力は一般に距離とともに指数関数的に減少するという物理的な前提に基づき、r最大– r分 適切に選択することで、元の分子における環境サブグラフ外の原子と勾配サブグラフ内の原子間の相互作用は無視できるようになります。そのため、学習時には元の分子ではなく環境サブグラフが入力として使用され、損失関数の計算には勾配サブグラフのみが使用されるため、分子構造データのスケールが適切にバランスされ、モデルの相互転送能力が向上します。
* 原子埋め込み拡張: より洗練された原子表現を実現
この研究は元素周期表に基づいています。同じ要素に対して複数の学習可能な離散埋め込み表現を拡張語彙として導入し、これは、原子が位置する規則的な部分構造を捉えるために使用されます。UniSimは、単純なグラフニューラルネットワークに基づいて、各原子の近傍情報を統合し、原子に対応する拡張語彙における各埋め込み表現の確率を取得し、重み付け和によって原子の拡張埋め込み表現を取得します。この表現は、原子レベルの精度と特定の分子種内の規則的なサブ構造とのバランスをとり、効率的で詳細な原子表現を実現します。
* マルチヘッドハイブリッド事前学習:異なる分子状態とラベル分布を持つデータのハイブリッド学習
UniSim は、定常状態と非定常状態の分子構造を共同で学習するために、次の方法を使用します。定常状態データの場合、この記事ではノイズ除去事前トレーニングパラダイムを使用してノイズの多いデータをノイズ除去し、原子の表現を学習します。非定常状態データの場合、モデルは保存力場を直接学習し、異なる力場パラメータが異なる出力ヘッドに対応するため、異なるラベル分布によって発生するエラーを回避できます。
本稿では、SO(3)同値性を満たす基本的なグラフニューラルネットワークモデルとしてTorchMD-NETを用いる。上記の主要な事前学習手法に基づき、大規模なマルチソース 3D 分子データの事前トレーニングが完了し、統一された原子表現モデルの効率的な構築が実現されました。
ベクトル場モデル:軌跡から長時間の状態遷移を学習する
従来の分子動力学シミュレーションは、数フェムト秒の積分ステップによって制限されており、タンパク質の折り畳みなどの長期的な動作を効率的にサンプリングすることが困難です。 UniSim は確率的補間フレームワークを採用し、事前トレーニング済みの全原子表現モデルの後に幾何学的ベクトル パーセプトロンをベクトル フィールド モデルとして接続します。このモデルは、長い時間ステップで分子状態間の転送ベクトル場を学習することにより、エンドツーエンドの時間粗化ダイナミクスモデリングを実現します。
学習中は、実際の動力学軌道において、所定の時間ステップだけ離れた分子構造のペアを学習サンプルとして選択し、補間経路にランダム摂動を導入し、速度場(velocity)とノイズ除去(denoiser)を共同学習することで、連続時間における軌道生成を実現します。従来の数値積分と比較して、UniSimはシミュレーション効率を大幅に向上させ、従来のシミュレーションにおける時間スケールのボトルネックを打破することができます。
力誘導核:複雑な化学環境への迅速な適応
異なる溶媒、温度、圧力条件下での分子動力学は異なるポテンシャルエネルギー面を持ち、生成されるコンフォメーションの分布に大きな影響を与えます。この目的のために、UniSim は、軌道サンプリングをガイドするためにランダム差分フレームワーク上に仮想中間力場を定義する力ガイダンスカーネルを導入します。この中間力場は、生成パスの両端(初期状態と最終状態)における実際の MD 力場と同等であり、物理的な事前条件と高い整合性を持つように設計されているため、生成されたコンフォメーションはターゲット力場におけるボルツマン分布とより整合性が高くなります。
中間力場をフィッティングすることにより、UniSim は事前トレーニング済みモデルとベクトル場モデルのパラメータを変更する必要がなくなります。ターゲット力場が新しい化学環境に効率的に適応するには、プラグ可能な力誘導カーネルを学習するだけで済みます。モデルの一般化および移行機能を効果的に強化します。
実験的検証:複数の分子タイプ
UniSimのさまざまな分子タイプにおける汎用性を検証するために、研究者らは、小分子、ペプチド、タンパク質の 3 種類の分子を含む、順方向シミュレーション タスクで複数の分子タイプからのデータを体系的に評価しました。この実験では、時間粗化動力学シミュレーションも実行するこの分野のディープラーニング モデルと比較することにより、統一された原子表現がモデルの分子状態の理解とクロスモーダル一般化機能の向上に役立つかどうか、また、力誘導核の関与がターゲット力場下で生成される配座の合理性や分布の類似性などの主要な指標におけるモデルのパフォーマンスにどのように影響するかを調査することを目的としています。
結果は、UniSim がすべての分子タイプにおいて総合的な優位性を達成したことを示しています。分布類似性において良好なパフォーマンスを示し、主要なコンフォメーション合理性指標(Val-CA)において大幅な改善が見られました。ただし、順方向シミュレーション生成タスクでは、軌跡上の各コンフォメーションが自己回帰によって生成されており、累積誤差が非常に大きいため、コンフォメーションの合理性を向上させるのは非常に困難です。
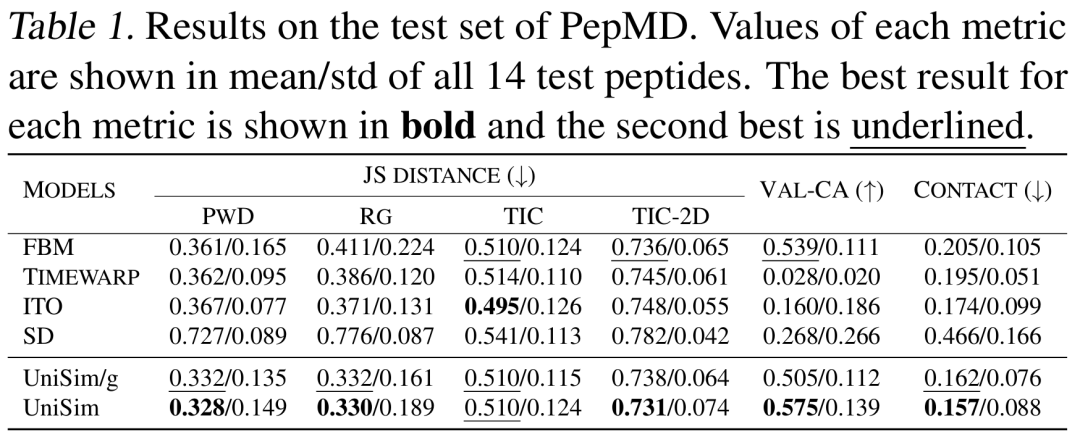
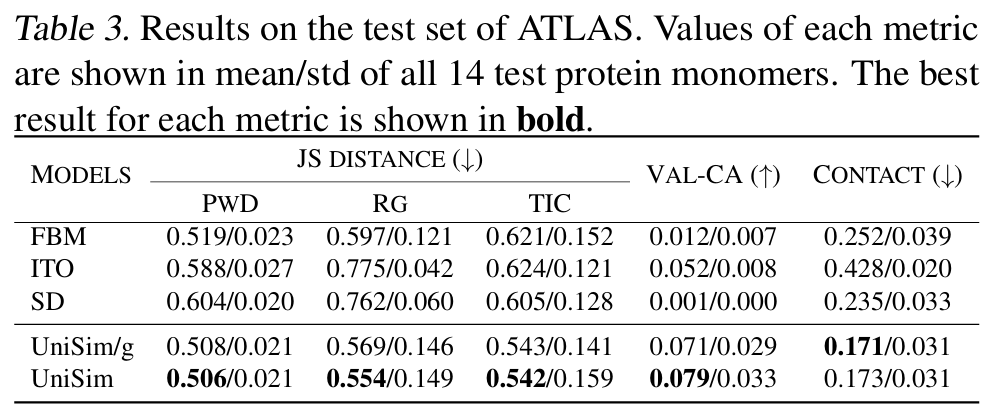
ペプチドとタンパク質の順方向シミュレーションタスクでは、UniSimは、FBM、ITO、SDなどの既存手法と比較して、分布類似性(TIC-2D)、構造合理性(VAL-CA)、接触マップエラー(CONTACT)などの指標において先行しています。特に、フォースガイドコアの導入後、UniSimは分布類似性などの指標では従来のレベルを維持しながら、主要なコンフォメーション合理性指標において大幅に改善しました。同時に、複雑なタンパク質系において、UniSimはわずか数百ステップの順方向シミュレーションでエネルギー障壁を飛び越え、複数の準安定状態をカバーできるため、巨大バイオ分子の効率的なシミュレーションの新たな方向性を切り開きます。
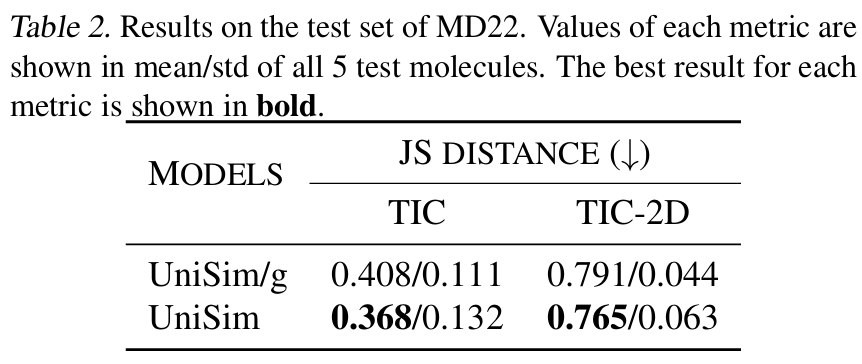
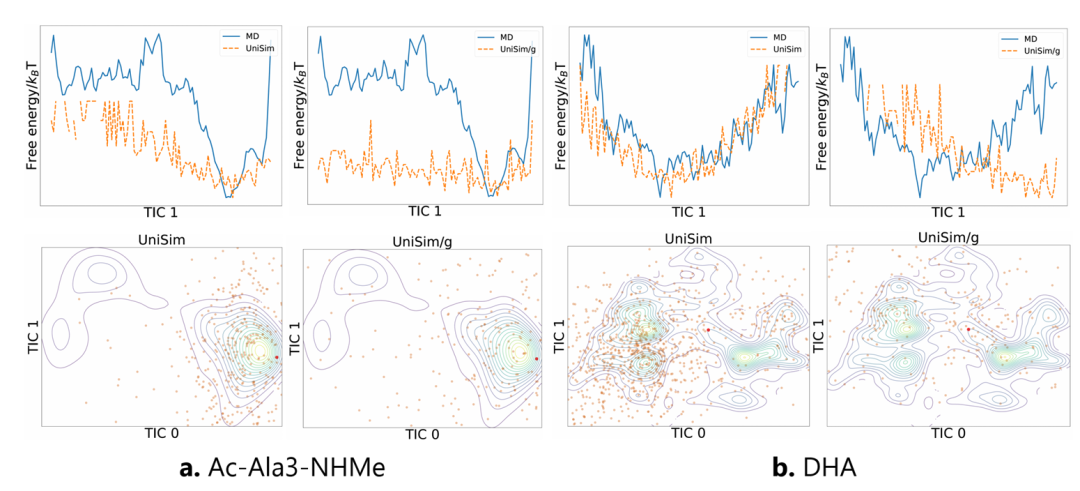
アラニンジペプチドのケーススタディ
さらに、長期分子動力学シミュレーションにおける UniSim の安定性を調査するために、研究者らは古典的なアラニンジペプチドのシステムでモデルを微調整し、100,000 ステップの長期シミュレーションを実行しました。MD 結果と比較することにより、UniSim は 5 つの既知の主要な準安定状態を正常に再現しました。動的プロセスにおけるアラニンジペプチドの自由エネルギーランドスケープが正確に復元され、長期シミュレーションにおけるモデルの安定性と物理的一貫性が完全に検証されました。
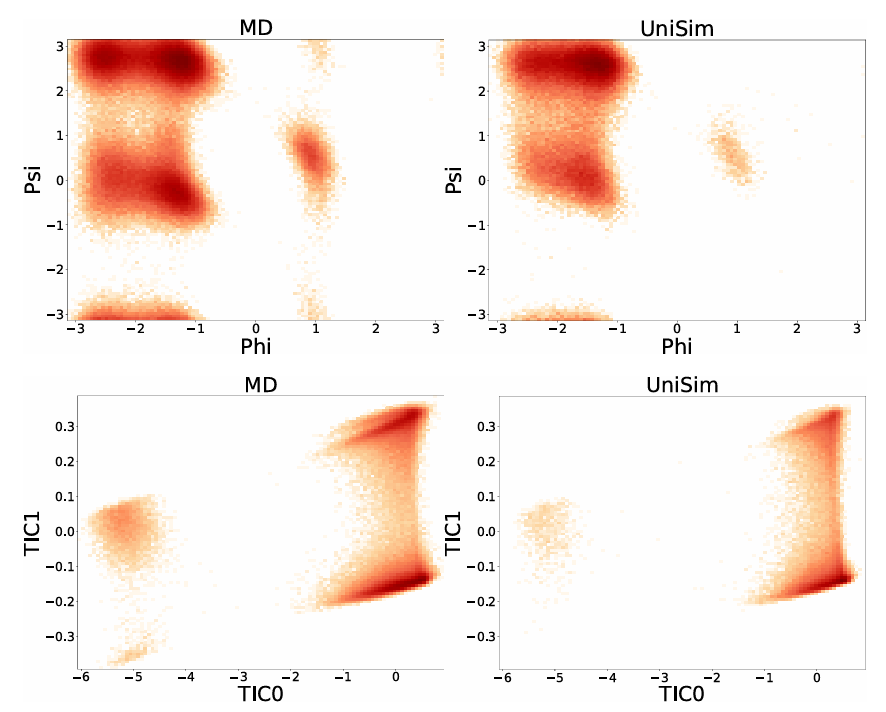
見通し
UniSim は、分子タイプと化学環境全体にわたって統一された時間的粗粒度ダイナミクスシミュレーションを実現する最初のフレームワークです。これは、創薬、タンパク質設計、その他の分野におけるディープラーニングの広範な応用に向けた実現可能な道筋を示しています。研究者らはまた、今後、以下の方向性をさらに探求できると指摘しています。
* 生成されたサンプルの有効性を向上させる、より効率的なクロスモーダルコンフォメーション最適化メカニズム。
* 複雑な生体物理学的メカニズムを明らかにするためのより長い時間スケールでの軌跡モデリング。
* 分子間相互作用に焦点を当てて、複雑なシステムの動的なメカニズムを探ります。








