Command Palette
Search for a command to run...
La Prédiction De La Structure De l'ARN Rivalise Avec AlphaFold 3 ! Une Équipe De Virginia Tech Propose RNAbpFlow, Totalement Indépendant Des Informations évolutives.

En novembre 2020, AlphaFold 2 s'est fait connaître lors du concours international de prédiction de structures protéiques CASP14. Grâce à sa précision quasi expérimentale dans la prédiction de la structure tridimensionnelle des protéines, AlphaFold 2 a non seulement résolu le problème du repliement des protéines qui hantait la communauté biologique depuis un demi-siècle, mais a également valu à ses deux créateurs le prix Nobel de chimie quatre ans plus tard, grâce à ses résultats exceptionnels. Cependant, le succès d'AlphaFold 2 n'était que le début. Tel un catalyseur, il a bouleversé le domaine de la biologie structurale, déclenchant une vague de recherches utilisant l'intelligence artificielle pour analyser la conformation spatiale des macromolécules biologiques.
Inspirés par la découverte révolutionnaire d'AlphaFold 2 dans le domaine des protéines, de grands espoirs reposent également sur l'utilisation de l'intelligence artificielle pour résoudre le problème de la prédiction de la structure tridimensionnelle de l'acide ribonucléique (ARN).Cependant, les algorithmes dominants actuels se heurtent encore à plusieurs obstacles pratiques inévitables lors de leur mise en œuvre :Premièrement, la plupart des modèles de prédiction d'ARN basés sur les Transformers s'appuient fortement sur les informations de séquences évolutives dominantes fournies par les alignements de séquences multiples (MSA) ou sur les informations de séquences homologues obtenues indirectement à partir de modèles de langage biologique. Cependant, la nature isostructurale de l'appariement des bases de l'ARN rend difficile la génération de résultats MSA fiables et de haute qualité. Deuxièmement, la plupart des méthodes courantes n'exploitent pas pleinement les informations clés de l'appariement des bases dans la structure secondaire de l'ARN, qui est le facteur déterminant de la morphologie de repliement tridimensionnelle de l'ARN. Troisièmement, les molécules d'ARN possèdent une flexibilité conformationnelle naturelle et existent sous de multiples conformations spatiales stables. Les algorithmes existants ne produisent généralement que des résultats statiques uniques et ne peuvent pas reconstruire l'ensemble des conformations dynamiques réelles de l'ARN.
Pour relever les défis susmentionnés, le professeur Debswapna Bhattacharya et son étudiant Sumit Tarafder de Virginia Tech ont collaboré pour développer un modèle de correspondance de flux SE(3)-équivariant, RNAbpFlow, basé sur des conditions de séquence et de paire de bases.Ce modèle peut générer un ensemble complet de conformations d'ARN atomiques uniquement à partir des séquences de nucléotides d'ARN et des informations d'appariement de bases, sans nécessiter d'informations évolutives telles que des alignements de séquences multiples ou des modèles homologues.Cette méthode remédie à plusieurs lacunes des méthodes existantes de prédiction de la structure de l'ARN basées sur l'IA. De nombreuses expériences comparatives ont démontré que l'introduction de l'appariement des bases comme contrainte améliore significativement la précision des prédictions du modèle. Pour les deux tâches principales que sont l'échantillonnage de la topologie de l'ARN et la modélisation de la conformation 3D, RNAbpFlow surpasse les méthodes courantes.
Il convient de mentionner que, lors du test comparatif entre RNAbpFlow et AlphaFold 3, un ensemble de données impressionnantes a révélé le résultat final du duel :Dans l'expérience de test à l'aveugle CASP16, RNAbpFlow a pu reconstruire avec précision la topologie de repliement globale de la grande majorité des 14 cibles d'ARN (≤200 nt), dont 12 répondaient aux critères de prédiction qualifiés.En revanche, dans les mêmes conditions expérimentales, seules 8 structures cibles prédites d'AlphaFold 3 répondaient aux critères de correspondance avec la conformation naturelle.
Les résultats de recherche pertinents, intitulés « RNAbpFlow : correspondance de flux SE(3) augmentée par paires de bases pour la génération de structures 3D d'ARN conditionnelles », ont été publiés dans Nature Methods.
Points saillants de la recherche :
- Le modèle RNAbpFlow proposé est un modèle d'appariement isofluidique SE(3) basé sur des conditions de séquence et de paires de bases, qui remédie à plusieurs lacunes des méthodes de prédiction de structure tridimensionnelle de l'ARN existantes.
- Trois innovations majeures ont été proposées : l'entrée conditionnelle d'appariement de bases à trois canaux, la caractérisation du centre de base des nucléosides et les pertes auxiliaires spécifiques à l'appariement de bases (bp2D et bp3D), qui ont considérablement amélioré la fidélité de l'appariement de bases et la structure tridimensionnelle.
- Nous proposons une stratégie d'ensembles d'entraînement et de test indépendants et non répétitifs, et utilisons plusieurs benchmarks de référence ainsi que des tests à l'aveugle CASP pour la validation. Les performances globales surpassent celles des algorithmes courants existants.

Adresse du document :
https://www.nature.com/articles/s41592-026-03128-4
La déduplication indépendante des ensembles d'entraînement et de test garantit une évaluation juste et équitable.
Pour construire le modèle auto-développé RNAbpFlow et garantir l'authenticité et la fiabilité de ses résultats d'évaluation, cette étude utilise des ensembles d'entraînement et de test indépendants sans duplication de contenu et attribue des pondérations de modèle spécifiques à différents points de référence de test, plutôt que d'utiliser un seul modèle par défaut tout au long du processus, afin d'éviter que les données d'entraînement ne soient mélangées au processus d'évaluation et n'entraînent une distorsion des résultats.
En ce qui concerne le développement du modèle et la validation interne,Cette étude utilise le jeu de données RNA3DB. Ce jeu de données est particulièrement adapté à l'entraînement de modèles d'apprentissage profond et à l'évaluation comparative interne, grâce à sa non-redondance aux niveaux de la séquence et de la structure. Dans cette étude, le jeu de données utilisé est la version extraite de la Protein Database (PDB) le 26 avril 2024, et le schéma de partitionnement entraînement-test décrit dans l'article original a été appliqué pour sélectionner des séquences d'ARN représentatives pour les expériences.
Afin de garantir que l'ensemble de données ne contienne que des structures d'ARN naturelles de haute qualité et des informations précises sur l'appariement des bases, l'expérience a fait l'objet d'un filtrage de contrôle qualité multicouche. Ce filtrage comprenait la suppression des structures ne comportant qu'un seul atome par nucléotide et des structures mélangées à des résidus protéiques ; la troncature des séquences expérimentales continues pour corriger les mésappariements entre les séquences FASTA et les structures tridimensionnelles, préservant ainsi l'intégrité des appariements de bases ; et la suppression des séquences sans appariement de bases dans les structures naturelles. Après extraction des informations d'appariement de bases à l'aide de RNAView, filtrage des chaînes d'ARN non appariées continues d'une longueur ≥ 20 nucléotides et limitation de la longueur des séquences à 30-200 nucléotides, un ensemble d'entraînement pur contenant 560 séquences d'ARN (les séquences ne correspondant pas à la famille Rfam ont été supprimées afin de réduire davantage le risque de fuite de données) et un ensemble de test pur contenant 48 séquences ont finalement été obtenus.
Il convient de mentionner que cette partie du partitionnement des données suit intégralement la division non redondante de RNA3DB, et qu'il n'y a aucun chevauchement de séquences ou de structures entre les ensembles d'entraînement et de test, afin d'éviter les fuites de données à partir de la racine.
Dans l'expérience comparative de RNAbpFlow et RNAJP,L'étude a utilisé un ensemble de données de 22 séquences d'ARN contenant des connecteurs à trois voies, similaires à ceux utilisés dans l'étude RNAJP, puis a procédé à un criblage. Les chercheurs ont d'abord supprimé les structures multimères de cet ensemble de données, puis ont rapidement comparé les séquences restantes avec l'ensemble d'entraînement nettoyé mentionné précédemment afin d'éliminer les redondances et les doublons. L'ensemble de données final traité a été réduit à 12 séquences.
En ce qui concerne la compatibilité avec les compétitions internationales CASP15 et CASP16,Afin de respecter scrupuleusement les règles de test à l'aveugle du concours et de garantir une comparaison équitable avec les autres algorithmes, l'étude n'a pas repris directement le partitionnement des ensembles d'entraînement et de test proposé par RNA3DB. Elle a plutôt réorganisé et planifié deux nouveaux ensembles d'entraînement totalement disjoints et non chevauchants à partir de RNA3DB.
Pour CASP15, le premier lot d'ARN à tester n'ayant été rendu public qu'en mai 2022, les chercheurs ont uniquement inclus, lors de la conception du nouvel ensemble d'entraînement, les séquences d'ARN téléchargées dans la base de données PDB avant avril 2022 (toutes issues de RNA3DB). Ceci a permis de garantir que l'entraînement du modèle excluait totalement les ARN testés lors de CASP15, respectant ainsi scrupuleusement les règles de test à l'aveugle. Au final, l'ensemble d'entraînement comprenait 731 séquences d'ARN, d'une longueur variant de 30 à 784 nucléotides. L'ensemble de test utilisé pour l'évaluation du modèle était composé de 6 ARN naturels et de 4 ARN synthétiques issus de l'ensemble de référence de CASP15.
Pour CASP16,L'ensemble d'entraînement utilisé dans l'expérience était composé de 994 structures PDB résolues expérimentalement et de 2 170 structures de prédiction à haute confiance utilisées pour l'augmentation des données de distillation croisée, pour un total de 3 164 échantillons.Le rapport entre le nombre de structures PDB et le nombre de données distillées croisées dans chaque lot d'entraînement est d'environ 1:2,2.
Pour la même raison, le premier lot d'ARN à tester dans le cadre de CASP16 a été publié en mai 2024. Les chercheurs ont donc fusionné toutes les données éligibles de RNA3DB en un seul ensemble d'entraînement, incluant uniquement les structures PDB téléchargées au plus tard le 6 avril 2024. Après une sélection rigoureuse, 994 séquences d'ARN et leurs structures tridimensionnelles mesurées correspondantes ont été obtenues. Les données de test proviennent de 28 ARN dont les structures tridimensionnelles expérimentales sont actuellement disponibles dans CASP16.
Les données utilisées pour construire l'ensemble de données de distillation croisée proviennent de l'ensemble de données bpRNA-1m(90), principalement pour explorer les effets de l'augmentation des données. L'ensemble de données bpRNA-1m(90) contient 28 370 séquences d'ARN (et leurs structures secondaires correspondantes). Après suppression des redondances et limitation de la longueur des séquences à 30-200 nucléotides, et après plusieurs extractions à l'aide de l'outil de clustering MMseqs2,Le résultat final a été un ensemble de données de distillation croisée de 2 170 structures d'ARN à haute confiance.
Cadre de génération conditionnelle basé sur la correspondance de flux
RNAbpFlow est un modèle d'appariement isofluidique SE(3) basé sur les conditions de séquence et d'appariement de bases (comme illustré dans la figure ci-dessous). Dérivé du modèle de génération de structures protéiques FrameFlow, il adopte la méthode de représentation des nucléotides proposée par NuFold, représentant chaque nucléotide de la séquence d'ARN par un cadre rigide. Utilisant la séquence nucléotidique et les informations d'appariement de bases comme contraintes, il prédit les angles dièdres par échantillonnage itératif, réduisant progressivement tous les atomes à leur structure tridimensionnelle finale et complète d'ARN, avec la conformation correcte du repliement du ribose. En bref,Les principaux avantages de RNAbpFlow peuvent être résumés en deux points : « conditions d'entrée » et « paradigme du modèle ».
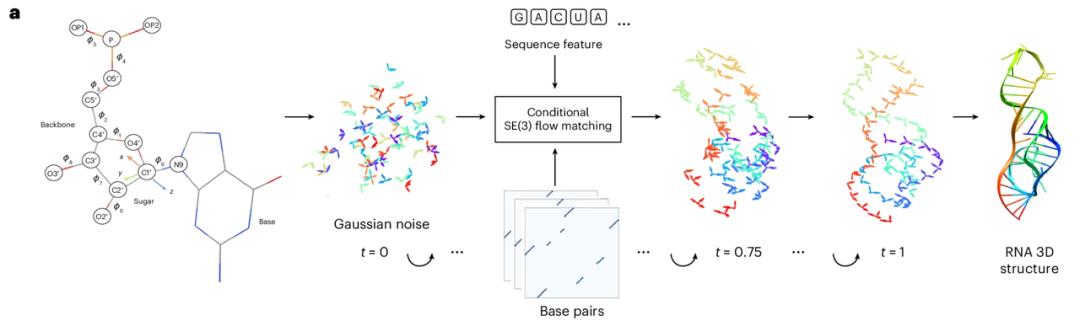
Concernant les « conditions d'entrée »,RNAbpFlow peut générer de manière conditionnelle des structures d'ARN tridimensionnelles sans aucune information de MSA ou de modèle homologue, en s'appuyant uniquement sur les informations de séquence et d'appariement de bases.
Premièrement, pour la séquence, une séquence d'ARN de longueur L est utilisée comme entrée, et les nucléotides sont représentés par un codage one-hot, c'est-à-dire qu'un vecteur binaire à quatre éléments correspond aux quatre types de nucléotides (A, U, C et G). Deuxièmement, pour le traitement des informations d'appariement de bases lors de la phase d'apprentissage (qui constitue l'innovation majeure), trois outils – RNAView, MC-Annotate et DSSR – sont utilisés pour extraire des annotations d'appariement bidimensionnelles à partir de la structure tridimensionnelle naturelle résolue expérimentalement. Ces annotations sont représentées par trois matrices binaires L × L indépendantes. Afin de capturer pleinement les différentes caractéristiques d'appariement de bases, classiques et non classiques, l'étude ne corrige pas uniformément les annotations contradictoires obtenues par les trois outils. Au lieu de cela, les trois matrices binaires sont directement concaténées en un tenseur L × L × 3, qui sert d'entrée au terme de biais dans la structure du réseau de débruitage, fournissant ainsi des informations sur les caractéristiques d'appariement dans trois canaux indépendants.
De plus, pour les scénarios d'échantillonnage et d'inférence des cibles de test des compétitions CASP15 et CASP16, en l'absence de valeurs réelles pour les appariements de bases naturels, les expériences ont utilisé des matrices de prédiction d'appariement dépendantes de la séquence, générées par trois outils de prédiction de structure 2D d'ARN : IPKnot, SPOT-RNA et RibonanzaNet. Ces trois outils prennent en charge la prédiction d'appariement de bases basée sur l'identification de pseudonœuds, et leur sélection s'est fondée sur leurs performances d'échantillonnage sur des cibles d'ARN naturelles dans CASP15. Par ailleurs, RNAbpFlow est très polyvalent et permet aux utilisateurs de définir trois ensembles de matrices d'appariement en entrée. Si un seul ensemble de matrices d'appariement personnalisées est disponible, il peut être dupliqué trois fois pour correspondre au format d'entrée 2D à trois canaux requis par le réseau.
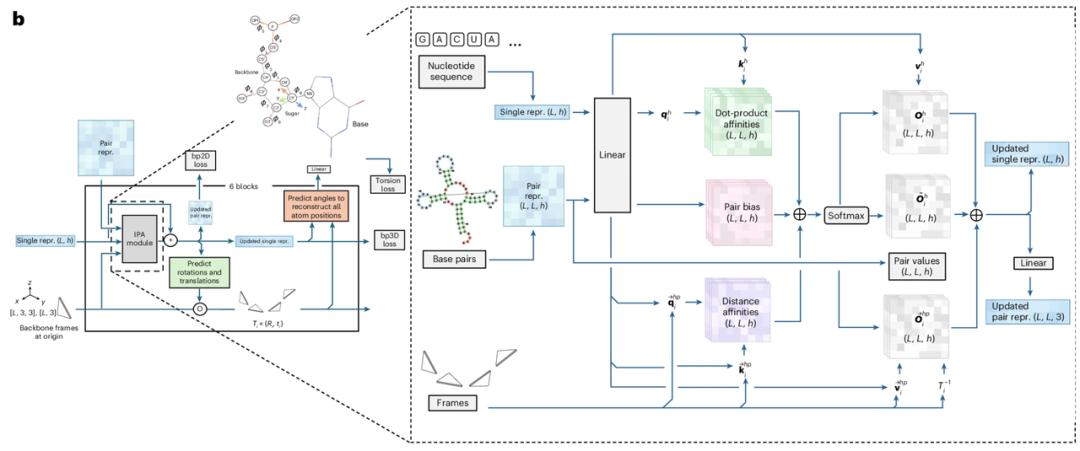
En termes de « paradigme de modèle »,La clé réside dans la « correspondance de flux ». La correspondance de flux est un type de modèle génératif profond dont l'objectif principal est d'apprendre un champ de vitesse (ou champ de flux) correspondant au flux de probabilité de la distribution des données. Ce faisant, elle transforme des distributions simples, comme les distributions gaussiennes, en la distribution de données complexe requise pour la cible dans un espace de grande dimension. La correspondance de flux apprend directement ce champ de vitesse pour décrire le mouvement des points d'échantillonnage migrant d'une distribution simple vers la distribution cible, sans perturber complètement la distribution des données originales. En intégrant des équations différentielles ordinaires sur le champ vectoriel appris, la correspondance de flux peut générer des trajectoires de migration plus simples pour approcher la cible. Comparée aux modèles de diffusion, la correspondance de flux peut améliorer considérablement la vitesse de calcul pour la génération d'échantillons à grande échelle.
Dans cette étude, la méthode d'appariement de flux vise à apprendre un champ vectoriel paramétré Ut. Ce champ vectoriel représente une application lisse et variable dans le temps, à partir de laquelle des équations différentielles ordinaires sont générées pour décrire la relation de transformation entre deux distributions : la distribution p₀(T₀) (repères bruités) et la distribution p₁(T₁) (repères de référence réels). Pour apprendre cette application, les chercheurs ont entraîné un réseau de neurones paramétré Vθ(Tₜ, t), qui prédit le champ vectoriel à partir du repère de référence réel bruité Tₜ à l'instant t. Cette partie du réseau est conçue en référence à FrameFlow, utilisant les modules structurels d'AlphaFold 2 comme architecture de base.
Concernant les paramètres d'entraînement spécifiques, l'expérience a été menée en utilisant le framework PyTorch-Lightning pour entraîner le modèle, en employant l'optimiseur Adam avec un taux d'apprentissage de 0,0001. Le processus d'entraînement distribué s'est déroulé sur 8 GPU NVIDIA H100 pendant 1500 itérations.
Les performances critiques surpassent celles d'AlphaFold 3.
Pour évaluer les performances d'échantillonnage de RNAbpFlow, l'étude l'a d'abord comparé à RNAJP, une méthode d'échantillonnage de structure d'ARN tridimensionnelle basée sur des simulations de dynamique moléculaire à gros grains qui prend explicitement en compte les informations d'appariement de bases, couvrant l'appariement de bases non classique, les interactions d'empilement de bases et les interactions boucle-boucle à longue portée.
Les résultats expérimentaux sont présentés dans la figure ci-dessous.RNAbpFlow a surpassé RNAJP dans les deux indicateurs d'évaluation.Plus précisément, RNAbpFlow a obtenu un score élevé au test de différence de distance locale moyen (score lDDT) de 0,66, tandis que RNAJP n'a obtenu que 0,59. De même, en termes d'échantillonnage de la topologie globale, RNAbpFlow a obtenu un score moyen de modélisation de modèle (score TM) de 0,38, tandis que RNAJP a obtenu 0,32.
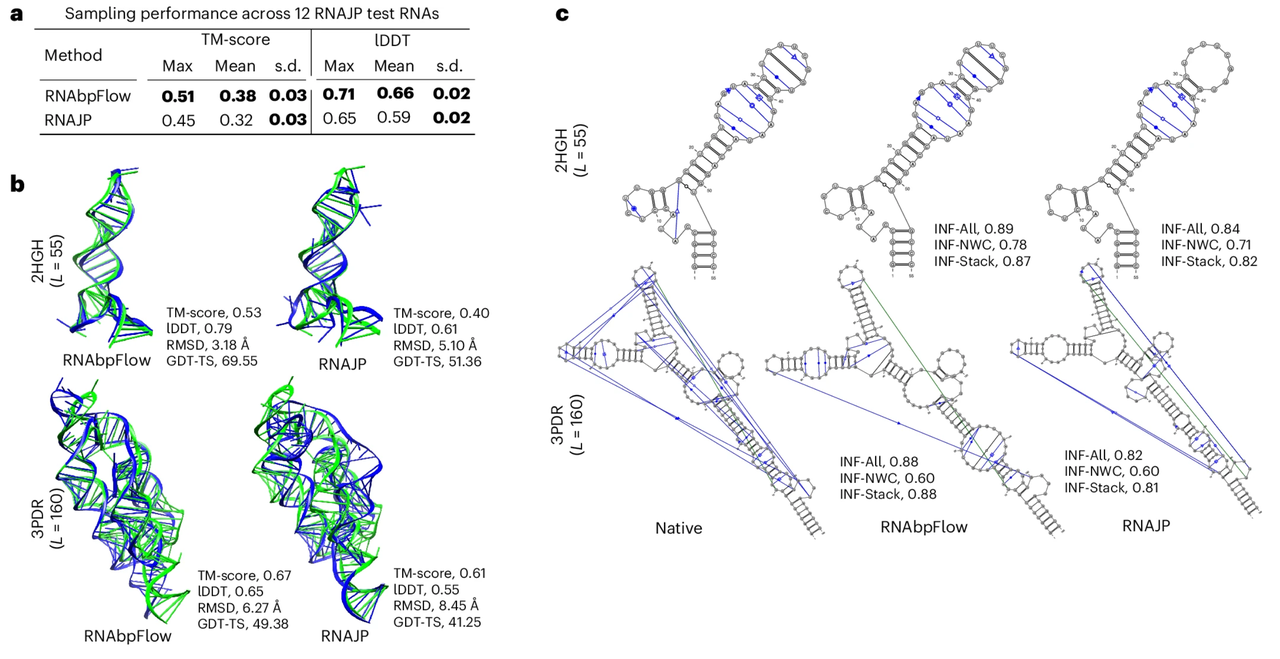
En ce qui concerne l’évaluation de la validité de l’échantillonnage, la structure de pliage correcte est déterminée sur la base du score TM.RNAbpFlow a détecté des cibles d'ARN aussi élevées que 66,671 TP3T.Le taux de détection des structures correctement repliées, basé sur les scores lDDT, était de 251 TP3T ; en revanche, les taux pour RNAJP n’étaient que de 41,671 TP3T et de 0 TP3T, respectivement. De plus, parmi les 12 000 conformations simulées générées par RNAbpFlow, 13,41 conformations TP3T présentaient des scores de modélisation de matrice supérieurs à 0,45, et 9,61 conformations TP3T présentaient des scores lDDT supérieurs à 0,7. En revanche, parmi les conformations simulées de RNAJP, seules 1,731 TP3T présentaient des scores de modélisation de matrice supérieurs à 0,45, et aucune conformation n’avait de score lDDT supérieur à 0,75.
Les résultats ci-dessus démontrent que RNAbpFlow surpasse non seulement RNAJP en termes de notation des structures optimales, mais atteint également un pourcentage plus élevé de conformations simulées de haute qualité.Cela met en évidence son efficacité dans les tâches d'échantillonnage double impliquant à la fois une topologie globale et une configuration locale.
L'équipe de recherche a ensuite comparé RNA bpFlow à plusieurs méthodes existantes sur l'ensemble de données de test à l'aveugle CASP15 (comme indiqué dans le tableau ci-dessous). Les résultats montrent que…Lorsque des informations réelles et précises sur l'appariement des bases naturelles sont saisies, les performances de modélisation et de prédiction de RNAbpFlow sont considérablement améliorées.Le score TM moyen a atteint 0,48, l'écart quadratique moyen de tous les atomes (RMSD) était de 7,77 et la fidélité du réseau d'interaction de paires de bases non-Watson-Crick (INF-NWC) était de 0,62 ; lorsque seuls les appariements de bases prédits par l'algorithme ont été utilisés, les trois indices étaient respectivement de 0,40, 10,70 et 0,48.
En comparaison, lorsque l'on fournit de véritables informations d'appariement de bases, le score TM a augmenté de 201 TP3T, le RMSD a diminué de 27,41 TP3T et l'INF-NWC a augmenté de 29,21 TP3T, soulignant l'importance d'informations d'appariement de bases de haute qualité.
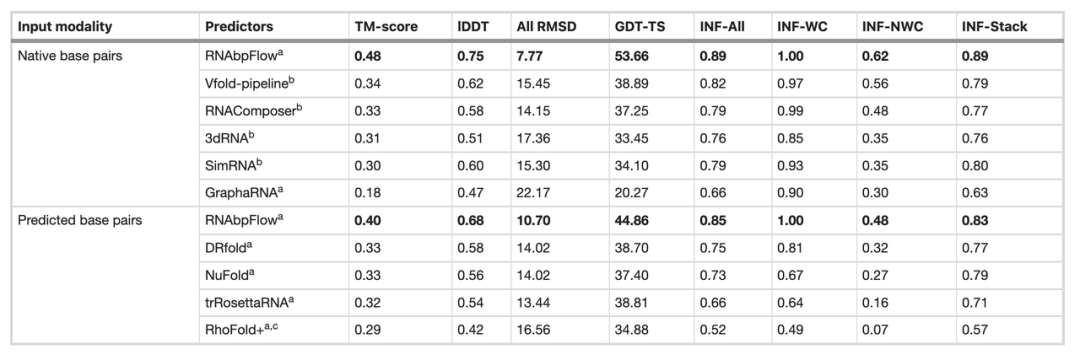
En revanche, d'autres méthodes n'affichent qu'une amélioration très limitée des performances, même avec des paires de bases naturelles réelles. Par exemple, Vfold, la méthode la plus performante, n'atteint qu'un score TM de 0,34, tandis que RNAComposer présente un RMSD minimal de 14,15. Ceci démontre la plus grande adaptabilité de RNAbpFlow, qui exploite efficacement les contraintes précises d'appariement de bases dans la modélisation générative profonde et repousse significativement les limites supérieures des performances prédictives des méthodes d'IA.
Pour vérifier la précision d'échantillonnage de RNAbpFlow sur les cibles de test à l'aveugle de CASP16, les chercheurs l'ont comparé aux deux modèles de prédiction les plus performants de la compétition CASP16. Les résultats (illustrés dans la figure ci-dessous) montrent que RNAbpFlow surpasse tous les algorithmes, y compris AlphaFold 3, en termes de score TM maximal moyen et de métrique lDDT pour 14 cibles prédites (≤ 200 nt). Parmi les conformations structurales générées par RNAbpFlow, au moins une conformation correctement repliée est présente dans 12 d'entre elles (85,711 TP3T) ; en revanche, AlphaFold 3 n'a atteint ce résultat que dans 8 conformations (57,131 TP3T), ce qui démontre la performance plus stable de RNAbpFlow.
Cependant, pour les cibles d'ARN de plus de 200 nucléotides, RNAbpFlow reste plus performant que NuFold, trRosettaRNA2 et DRfold2, mais moins performant qu'AlphaFold3. Cela s'explique par une fidélité de prédiction des appariements de bases significativement réduite pour les longues séquences.
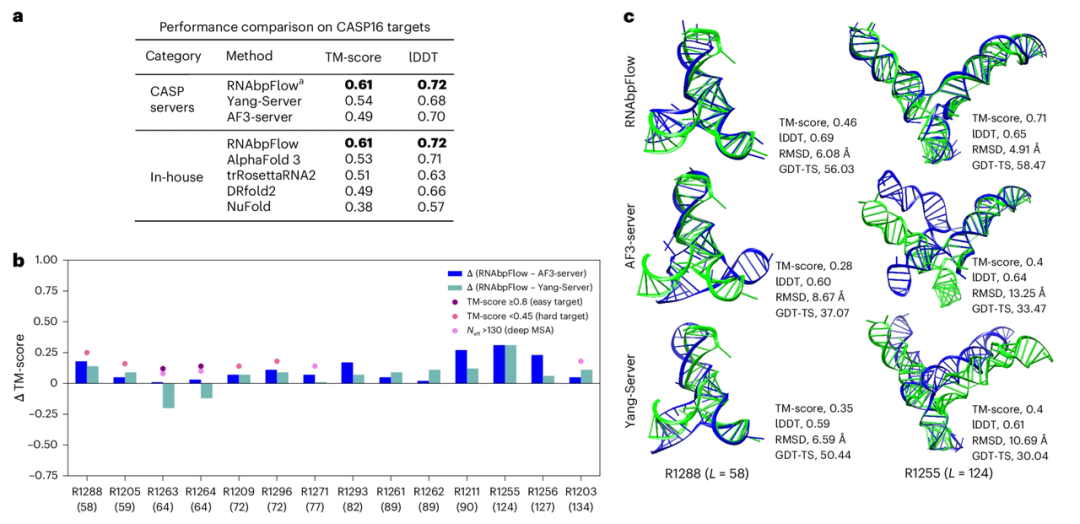
De plus, les expériences ont démontré que pour les cibles imprévisibles (score TM < 0,45 ; profondeur de séquence d'homologie effective MSA ≤ 130 ; signal évolutif faible), le modèle de prédiction de structure basé sur les contraintes d'appariement de bases surpassait nettement RNAbpFlow lorsque l'information évolutive était rare, mettant en évidence la supériorité de RNAbpFlow, capable de prédire la structure tridimensionnelle de l'ARN uniquement à partir des informations d'appariement de bases. Cependant, face à des cibles facilement prévisibles telles que R1263 et R1264, des données d'alignement multiple de séquences profondes et suffisantes ont permis aux deux modèles d'alignement, AF3-server et Yang-Server, d'égaler, voire de surpasser, RNAbpFlow, démontrant ainsi la forte dépendance des deux modèles aux informations d'alignement de séquences.
Derniers mots
En résumé, RNAbpFlow ne se limite pas à l'alignement multiple de séquences (MSA) ni à l'homologie structurale. Il peut générer directement des modèles tridimensionnels de structures d'ARN complètes, atomistiques et de bout en bout, à partir de la seule séquence et de l'appariement des bases. Grâce à une technologie de génération de conformations à grande échelle et de haute précision à l'échelle atomique, il pourrait ouvrir une voie très prometteuse pour l'étude de la dynamique conformationnelle de l'ARN.








