Command Palette
Search for a command to run...
À Partir De 220 Espèces De Bactéries Marines, Des Scientifiques Ont Reconstitué Le Système De Classification Microbienne Hétérotrophe À l'aide d'un Modèle À l'échelle Du Génome, Identifiant Huit Types De Flore métabolique.

Les forêts sont considérées comme les poumons de la Terre, tandis que l'océan en est le cœur. Les vastes océans abritent des dizaines de milliers de micro-organismes formant des communautés complexes qui régulent la transformation de la matière organique et pilotent les processus de fixation et de libération du carbone grâce à leurs spécialisations métaboliques uniques. Ces micro-organismes influencent profondément les cycles mondiaux du carbone, le changement climatique et la biodiversité marine. Parmi eux, les micro-organismes hétérotrophes marins agissent comme des « unités de purification » au sein de l'écosystème marin, assurant la fonction essentielle de dégradation de la matière organique et contribuant ainsi au maintien des cycles globaux de la matière et de l'équilibre écologique.
Longtemps, les micro-organismes hétérotrophes marins ont été classiquement classés en deux grandes catégories : les copiotrophes et les oligotrophes. Les premiers prospèrent dans les milieux riches en matière organique, tandis que les seconds survivent lentement dans les milieux pauvres en ressources. Cette dichotomie traditionnelle, utilisée depuis de nombreuses années, a certes contribué à la recherche biogéochimique, mais elle présente aussi des lacunes importantes : le taux de croissance ne peut être assimilé aux préférences d’utilisation des substrats ni aux niches métaboliques. De même qu’on ne peut classer les habitudes alimentaires humaines uniquement par la quantité de nourriture consommée, les préférences alimentaires sont un facteur déterminant du taux de décomposition de la matière organique et de la direction du cycle du carbone.
Pour remédier à ce problème, une équipe de l'Université de Californie du Sud, s'appuyant sur la base de données microbiennes océaniques (OMD), a analysé d'énormes quantités de génomes bactériens marins à l'aide de modèles métaboliques à l'échelle du génome (GEM). En quantifiant la sensibilité des micro-organismes à l'utilisation de 11 types de substrats organiques, ils ont finalement dépassé le cadre dichotomique traditionnel.Huit catégories de microbiote métabolique différencié ont été identifiées : une catégorie de microbiote eutrophe à croissance rapide, trois catégories de microbiote oligotrophe à croissance lente spécifique au substrat et quatre catégories de microbiote à croissance intermédiaire spécialisé au substrat.
Les résultats, intitulés « Définition des niches métaboliques des hétérotrophes microbiens marins », ont été publiés dans Science Advances.
Points saillants de la recherche :
* Rompant avec le cadre classique de la « dichotomie », elle ancre des niches métaboliques spécifiques aux microbes en se basant sur des stratégies métaboliques réelles et des préférences de substrat.
* Basée sur huit communautés microbiennes fonctionnelles, cette étude révèle systématiquement les schémas de croissance, les modèles de compétition pour les ressources et la distribution géographique mondiale des micro-organismes hétérotrophes marins, élucidant le mécanisme intrinsèque par lequel les micro-organismes pilotent le cycle du carbone marin.
* Combler les lacunes de la recherche concernant la participation des micro-organismes hétérotrophes marins au cycle global du carbone, et proposer des idées d'amélioration et des schémas de paramètres plus précis pour les modèles biogéochimiques.

Adresse du document :
https://www.science.org/doi/10.1126/sciadv.adz0537
Jeu de données : Couvrant 220 catégories différentes de bactéries marines
Cette étude est basée sur un ensemble de données à grande échelle de génomes microbiens marins, tiré de la base de données OMD hébergée sur la plateforme microbiomics.io.La base de données contient environ 35 000 génomes microbiens.Il comprend des génomes assemblés métagénomiquement, des génomes amplifiés à partir de cellules uniques et des génomes de souches isolées et cultivées artificiellement.
Cette étude a inclus uniquement les génomes bactériens présentant une intégrité supérieure à 80% et un taux de contamination inférieur à 5%. Ces deux ensembles de valeurs ont été calculés à partir des scores moyens obtenus respectivement avec les logiciels CheckM et Anvi'o. Par la suite, les chercheurs ont utilisé les métadonnées de la base de données OMD et le logiciel dRep pour éliminer la redondance des génomes, en appliquant un seuil d'identité nucléotidique moyenne (ANI) de 95%.
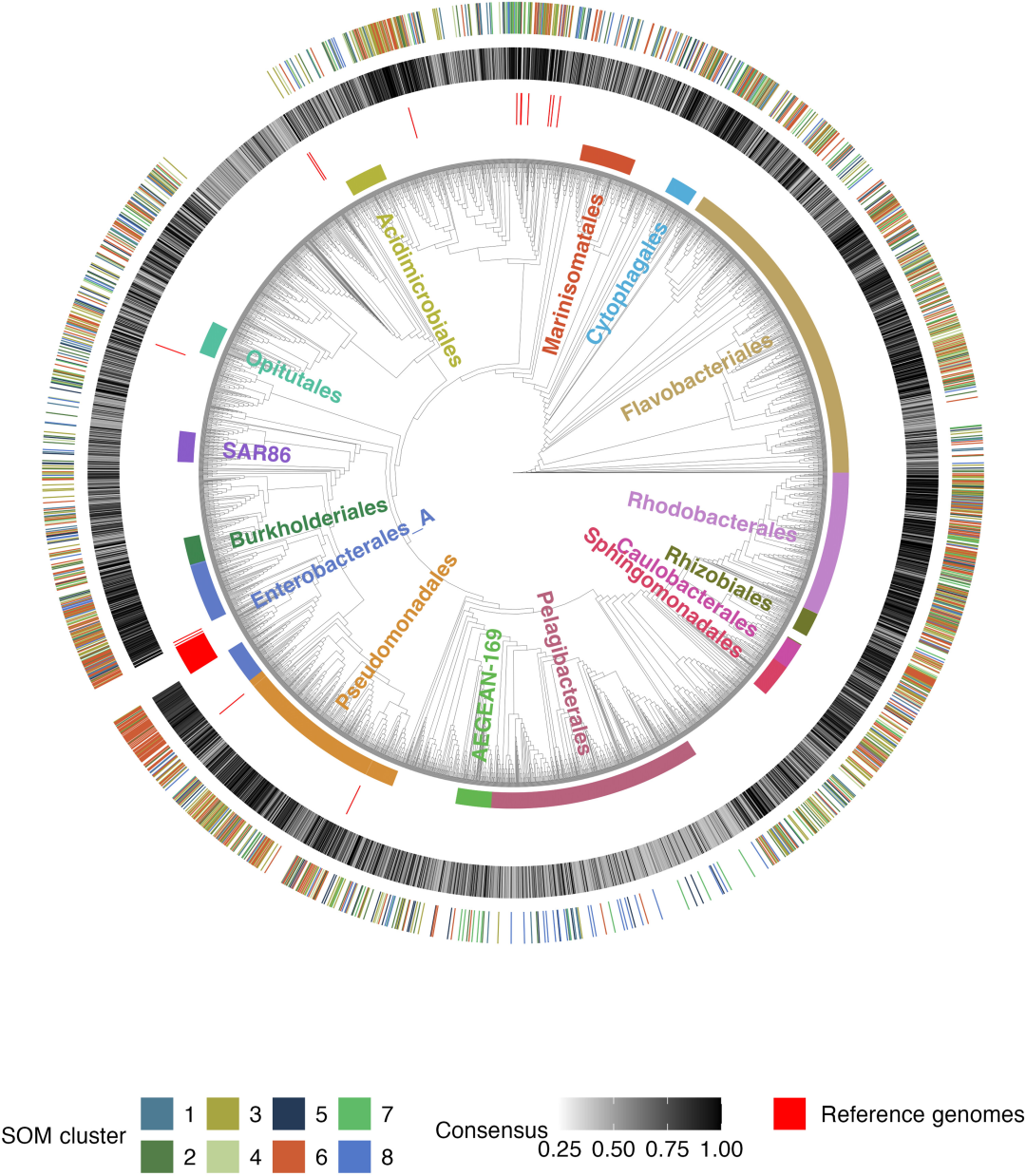
Après avoir éliminé 180 cyanobactéries autotrophes photosynthétiques (exogroupes),Au final, 3 738 génomes bactériens hétérotrophes de haute qualité ont été obtenus après élimination des redondances.Cet ensemble de données constitue la base de l'analyse pour cette étude. Il couvre 220 catégories différentes de bactéries marines, dont 14 groupes contiennent au moins 50 génomes.
Pour construire l'arbre phylogénétique, outre la conservation de 180 génomes de cyanobactéries comme groupes externes, 66 génomes bactériens de référence provenant de la base de données BiGG ont été ajoutés, portant le nombre total de génomes à 3 984. Huit génomes ont été retirés de l'arbre phylogénétique en raison d'une correspondance insuffisante des gènes cibles à copie unique.Au final, 3 976 plantes ont été utilisées pour construire les arbres de développement.L'ensemble des informations de classification du génome a été étiqueté sur l'arbre à l'aide des bases de données GTDB-Tk v2.1.0 et GTDB r214.
Les réseaux neuronaux de type carte auto-organisatrice sont utilisés pour classifier les niches métaboliques.
Afin de dépasser les limites du cadre « dichotomique » traditionnel,Cette étude intègre la génomique, la simulation métabolique contrainte et les techniques d'apprentissage automatique non supervisé pour construire un cadre analytique complet allant de l'information génétique à l'écotypage microbien.La modélisation métabolique, la quantification de la sensibilité au substrat et le regroupement des communautés microbiennes ont été réalisés de manière hiérarchique à l'aide de plusieurs types de mesures sur le terrain et d'ensembles de données environnementales mondiales.
Modélisation et contrôle de la qualité
Lors de la phase de modélisation, les chercheurs ont utilisé une stratégie de modélisation intégrée.Le logiciel CarveMe v1.5.1 a été utilisé pour construire 60 modèles métaboliques indépendants (ensembles de modèles) pour chacune des 3738 souches de bactéries hétérotrophes marines.
Plus précisément, le principe de modélisation du logiciel CarveMe repose sur une architecture de modèle métabolique général. En fonction de la présence ou de l'absence de chaque réaction biochimique dans les informations d'annotation du génome d'entrée, il attribue des pondérations à chaque étape réactionnelle, initialisant ainsi le modèle général et prédisant le modèle métabolique du génome correspondant. Cette étude a exploré de manière exhaustive le nombre de répétitions du modèle nécessaires pour couvrir le profil réactionnel d'un génome unique. Les résultats montrent que lorsque le nombre de modèles dans l'ensemble atteint environ 60, le nombre total de nouvelles réactions ajoutées se stabilise.Cela démontre qu'un ensemble de 60 modèles peut couvrir la grande majorité des combinaisons de modèles métaboliques possibles pour un seul génome.
Pour quantifier la qualité du modèle métabolique produit par CarveMe, l'étude a introduit un indice de score de cohérence C comme mesure d'évaluation, comme le montre la formule suivante :
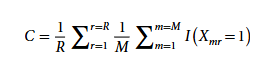
Dans la formule, Xmr représente la matrice de présence-absence de la réponse du modèle d'ensemble dans les M modèles construits indépendamment ; r désigne une réponse biochimique individuelle ; R est le nombre total de réponses dans l'ensemble des modèles ; et I est une fonction indicatrice permettant de déterminer si la réponse r est présente dans le m-ième sous-modèle d'ensemble. L'analyse ultérieure n'a conservé que les échantillons génomiques présentant un score de cohérence ≥ 0,8, soit un total de 1 578 génomes.
Évaluation de la stratégie métabolique
Les chercheurs définissent les stratégies métaboliques comme l'ensemble privilégié des substrats pour la croissance d'un organisme, et leur méthodologie consiste à interpréter ces stratégies à travers une série d'analyses de sensibilité.
Spécifiquement,Les chercheurs ont utilisé l'outil d'analyse de flux d'équilibre (FBA) du logiciel COBRApy v0.25.0 pour effectuer des tests de sensibilité de croissance sur le modèle CarveMe dans des conditions d'approvisionnement en substrat à la fois « substrat suffisant » et « substrat limité ». La condition « limitée par le substrat » est établie en réduisant le flux disponible d'une certaine classe de composés à 50% de la quantité que l'organisme absorberait dans des conditions « suffisantes en substrat ».
Pour quantifier les différences de besoins en substrat entre les différents modèles, un indice de coefficient de sensibilité S est proposé, comme indiqué ci-dessous :
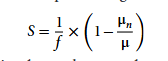
Dans la formule, μn représente le taux de croissance prédit en condition de limitation du substrat pour le type n, μ est le taux de croissance prédit en condition d'abondance du substrat, et f est le coefficient de limitation du substrat (fixé à 0,5 dans cette étude). Le coefficient de sensibilité S varie de 0 à 1. Si le taux de croissance calculé par le modèle diminue de 50% après une réduction de l'apport de substrat de 50%, alors ce type de substrat est considéré comme un facteur limitant total pour la croissance de l'organisme (sensibilité de croissance = 1). Si le taux de croissance modélisé reste inchangé, cela signifie que la croissance de l'organisme n'est pas affectée par l'apport de ce type de substrat (sensibilité de croissance = 0).
De plus, lorsque le rapport entre l'étendue de la restriction du substrat et l'étendue de la diminution du taux de croissance est ≥ 0,8 (S ≥ 0,8), le modèle est considéré comme ayant une sensibilité de croissance significative à de tels substrats.
Analyse de regroupement dans l'apprentissage automatique non supervisé
La partie de l'étude consacrée à l'apprentissage automatique a utilisé des cartes auto-organisatrices (SOM) pour délimiter les niches métaboliques. Les SOM sont un algorithme d'apprentissage automatique non supervisé qui peut réduire la dimensionnalité d'ensembles de données massifs de grande dimension à un espace de grille bidimensionnel avec une structure topologique.
Avant le regroupement, les chercheurs ont procédé à un criblage des données des 1 578 génomes obtenus précédemment et ont calculé les variances de sensibilité à la croissance de divers métabolites dans les 60 sous-modèles. Ils ont éliminé 100 génomes présentant une variance totale de sensibilité au substrat supérieure à 0,1, conservant ainsi 1 478 génomes. Au total, 88 680 ensembles de données valides (1 478 génomes x 60 modèles d'ensemble) ont été utilisés pour l'analyse de regroupement par cartes auto-organisatrices (SOM). Chaque point de données comprenait 11 indicateurs caractéristiques de sensibilité métabolique.
Concernant les paramètres expérimentaux, cette étude a utilisé le logiciel Kohonen v3.0.12 pour traiter les données de prédiction de flux de composés standardisées. 1 500 itérations ont été réalisées sur une grille hexagonale toroïdale 20 × 20 (la distance euclidienne standard étant utilisée pour caractériser la distance spatiale). Le taux d’apprentissage a été fixé à (0,025 ; 0,01) et le rayon du voisinage a été sélectionné parmi les valeurs par défaut du logiciel.
Après un entraînement suffisant, et en fonction de la cohérence des résultats de prédiction de la sensibilité des composés de croissance, l'algorithme de clustering K-means est adopté.La carte SOM a finalement été divisée en 8 groupes différentiels.
Après regroupement, afin d'évaluer les différences de taux de croissance maximal, l'étude a calculé le dCUB des 1 478 génomes à l'aide de gRodon pour classer les types de croissance en rapide et lente. La distribution géographique mondiale des huit communautés bactériennes a été validée à partir de 1 209 données métagénomiques issues de plusieurs jeux de données, dont Tara Oceans, BioGeoTraces et Malaspina, ainsi que d'un jeu de données ASV global de McNichol et al.
Huit groupes métaboliques ont été classés en fonction de la préférence pour le substrat et du taux de croissance.
L'étude présente des résultats expérimentaux variés qui non seulement valident les performances du modèle, mais surtout, brisent le cadre de la « dichotomie » traditionnelle, proposent une logique de classification totalement nouvelle et établissent la relation intrinsèque entre la préférence du substrat et la niche métabolique.
Résultats de validation du modèle
Des chercheurs ont validé la capacité du modèle CarveMe à reproduire les préférences de substrat lors d'expériences de culture à grande échelle portant sur les préférences de source de carbone de 186 micro-organismes marins. Plus précisément, ils ont construit un modèle CarveMe pour les génomes étudiés par Gralka et al. et mené des expériences in silico correspondantes à l'aide de l'analyse de flux métabolique (FBA) afin de tester la croissance de ces micro-organismes dans les mêmes conditions de source de carbone.
Les résultats montrent queComparativement aux données expérimentales de la littérature, les résultats de prédiction du modèle ont atteint une précision de 75,51 TP3T et une précision de 87,41 TP3T.Pour déterminer si ce résultat était significativement meilleur qu'une prédiction aléatoire, les chercheurs l'ont testé en effectuant une analyse bootstrap sur la prédiction aléatoire, et les résultats ont montré que la précision du modèle était significativement supérieure à celle du niveau aléatoire.
Résultats du typage de 8 types de bactéries
Sur la base de 1 478 séquences génomiques et de 11 indicateurs sensibles,L'étude a identifié huit communautés microbiennes métaboliques exprimées différemment grâce au regroupement SOM et les a classées en trois groupes principaux — rapide, moyen et lent — en fonction de leurs taux de croissance.Plus précisément (comme le montre l'image ci-dessous) :
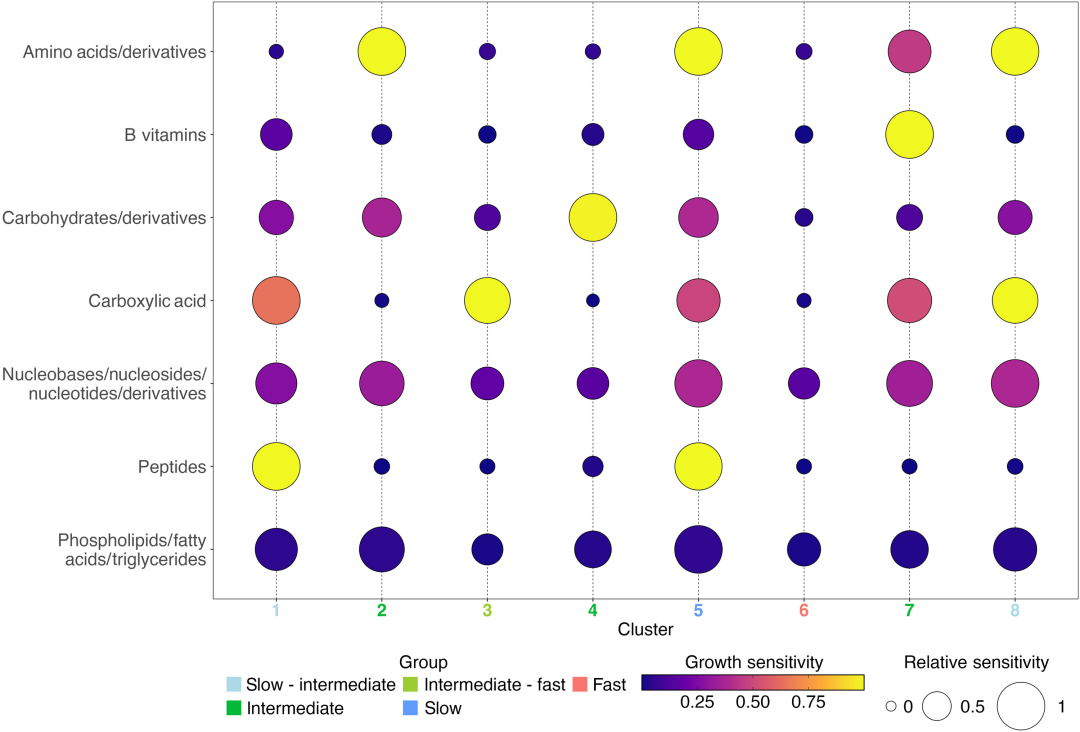
Comparaison de la sensibilité moyenne à la croissance de 8 groupes de SOM
Catégorie 1 : Microbiote eutrophe à croissance rapide (Cluster 6) : Il s’agit d’un microbiote eutrophe typique, dont le génome 79.5% prédit un taux de croissance maximal supérieur au seuil de croissance lente (dCUB < -0,08 correspond au seuil de croissance rapide ; une valeur de dCUB plus faible indique une croissance plus rapide). D’un point de vue taxonomique, les représentants typiques du Cluster 6 comprennent les Enterobacterales, les Flavobacteriales, les Rhodobacterales et les Pseudomonadales. Ce type de microbiote est peu affecté par les substrats ; l’absence de l’un des 11 composés testés n’a pas inhibé sa croissance.
Trois groupes bactériens oligotrophes à croissance lente et spécifiques à un substrat (clusters 1, 5 et 8) présentent un dCUB de -0,111. Le cluster 5 (61,81 TP3T) possède le taux de croissance maximal le plus faible, avec comme représentants typiques les Opitutales (Verrucomicrobiota) et les Pelagibacterales, dont l'enrichissement dans ce groupe atteint respectivement 4351 TP3T et 3621 TP3T.
Quatre groupes bactériens à croissance intermédiaire spécifiques au substrat (clusters 2, 3, 4 et 7) : ces groupes devraient présenter des taux de croissance significativement inférieurs à ceux du cluster 6, mais significativement supérieurs à ceux du cluster 5. Le cluster 3 a montré un taux de croissance significativement meilleur que les clusters 1 et 8. Chacun de ces quatre groupes bactériens à croissance intermédiaire a présenté une sensibilité de réponse de croissance à un seul type de composé : acides aminés, acides carboxyliques, glucides et vitamines B.
De plus, les caractéristiques des communautés microbiennes à croissance intermédiaire confirment une étude récente suggérant que les groupes microbiens hétérotrophes dominants dans le milieu marin subsuperficiel pourraient être des bactéries eutrophes à croissance lente. Cette découverte peut servir de base à la classification de ces micro-organismes en groupes fonctionnels métaboliques, comme par exemple la préférence du groupe 4 pour les glucides.
Derniers mots
En résumé, cette étude rompt avec le cadre « dichotomique » vieux de plusieurs décennies des niches eutrophes/oligotrophes et établit un système de classification des niches métaboliques en huit catégories basé sur l'essence de l'utilisation des gènes et des substrats, brisant ainsi le lien inhérent entre les cinq catégories et les fonctions physiologiques.
De plus, ce nouveau cadre de classification simplifie la structure complexe du vaste ensemble de micro-organismes hétérotrophes océaniques. À l'avenir, il pourra être intégré à des modèles biogéochimiques globaux, évitant ainsi le recensement individuel de dizaines de milliers de bactéries marines. Il permettra de déduire la dégradation de la matière organique marine et les variations du bilan carbone à partir de seulement huit paramètres fonctionnels, offrant ainsi un outil théorique inédit pour évaluer l'évolution du cycle du carbone océanique dans le contexte du réchauffement climatique.








