Command Palette
Search for a command to run...
Une Nouvelle Percée Dans La Recherche Biomédicale Sur Petits Échantillons : Une Équipe Allemande a Réalisé Une Augmentation Des Données Basée Sur Un Modèle d'IA Génératif, Réduisant Potentiellement Le Nombre d'animaux De Laboratoire Nécessaires De 30 À 50 Par TP3T.

Les « effets thérapeutiques efficaces » observés lors d’expérimentations animales peinent souvent à être reproduits lors d’essais cliniques, notamment en raison d’un échantillon insuffisant. De multiples contraintes, telles que les réglementations éthiques, les coûts des expérimentations et les conditions de recherche, aggravent encore le problème.La recherche biomédicale préclinique est souvent confrontée à des difficultés pour mener des expériences à grande échelle sur des animaux, ce qui entraîne directement une puissance statistique insuffisante.Les chercheurs sont incapables d'extraire de manière fiable les véritables signaux biologiques et sont très susceptibles d'obtenir des résultats faussement positifs, ce qui entrave sérieusement la transposition de la recherche fondamentale en applications cliniques.
Pour relever ce défi, les universitaires ont tenté d'intégrer les données de recherche en utilisant des méthodes telles que la méta-analyse et la fusion de données.Cependant, ces méthodes dépendent fortement de la comparabilité des protocoles expérimentaux, des indicateurs de détection et des procédures opérationnelles entre les différentes études.Son champ d'application pratique est extrêmement limité.
Ces dernières années, l'intelligence artificielle générative a offert une approche novatrice pour la recherche sur de petits échantillons : en apprenant la structure de distribution inhérente des données originales, elle génère des données synthétiques afin d'augmenter la taille de l'échantillon. Cependant, les modèles génératifs généraux présentent des lacunes importantes :Si les données originales contiennent des erreurs aléatoires, le modèle amplifiera davantage le bruit et générera un grand nombre de faux positifs, ce qui réduira la crédibilité des conclusions de la recherche.La question de la suppression de la propagation des erreurs lors de la génération de données est devenue un goulot d'étranglement majeur pour l'application de l'IA générative dans le domaine biomédical.
Pour remédier à ce problème crucial, une équipe de recherche conjointe de l'Université de Francfort et de l'Institut Fraunhofer pour l'ITMP a développé genESOM—Les modèles d'IA générative basés sur des cartes auto-organisatrices émergentes sont conçus spécifiquement pour les données biomédicales issues de petits échantillons.L'innovation majeure de ce modèle réside dans le découplage de l'apprentissage de la structure et du processus de génération des données. Il bloque la propagation des erreurs grâce à un ajustement de la dimensionnalité et introduit une variable de contrôle négative pour surveiller en temps réel la qualité de la génération des données. L'équipe de recherche a utilisé des données lipidomiques précliniques de la sclérose en plaques comme objet d'étude, en réduisant d'abord artificiellement la taille de l'échantillon jusqu'au seuil d'échec statistique, puis en utilisant genESOM pour réaliser une augmentation des données.
Les résultats confirment que cette méthode permet de récupérer efficacement des signaux biologiques clés perdus dans les données issues de petits échantillons, tout en contrôlant rigoureusement les faux positifs. Elle offre ainsi une nouvelle approche fiable pour la recherche biomédicale sur petits échantillons. De plus, dans le cadre de recherches exploratoires, ce modèle devrait permettre de réduire le nombre d'animaux d'expérimentation nécessaires d'environ 301 à 501, tout en préservant la reproductibilité et la validité scientifique des résultats.
Les résultats de recherche connexes, intitulés « Une IA générative basée sur un réseau neuronal auto-organisé avec contrôle intégré de l'inflation des erreurs améliore l'extraction efficace des connaissances à partir d'études précliniques avec une taille d'échantillon réduite », ont été publiés dans Pharmacological Research.
Points saillants de la recherche :
* Un mécanisme de contrôle d'erreur intégré basé sur les données supprime efficacement l'inflation des faux positifs, contrairement aux méthodes non contraintes telles que les GAN.
* Restauration réussie des principaux signaux lipidiques (tels que l'acide lysophosphatidique) après réduction de la taille de l'échantillon, sans augmentation du taux de faux positifs.
* Il peut réduire de 30 à 50 % la quantité de TP3T utilisée chez les animaux, en servant d'outil analytique complémentaire tout en tenant compte à la fois de la robustesse de la recherche et des principes éthiques des 3R.

Voir le document :
https://www.sciencedirect.com/science/article/pii/S1043661826000745
Jeux de données : des expériences complètes aux échecs statistiques sur petits échantillons
Les données de cette étude proviennent d'une étude préclinique animale sur la sclérose en plaques, publiée publiquement.L’étude a utilisé des souris SJL/J pour établir un modèle d’encéphalomyélite auto-immune expérimentale (EAE) récurrente-rémittente.L’étude vise à élucider les mécanismes des maladies neuro-inflammatoires et à valider l’efficacité thérapeutique du fingolimod, médicament approuvé.
Remarque : Le fingolimod est un modulateur des récepteurs de la sphingosine-1-phosphate qui peut interférer avec les voies de signalisation immunitaire en régulant le métabolisme des sphingolipides. C’est un médicament couramment utilisé dans le traitement clinique de la sclérose en plaques.
L'expérience a porté sur 26 souris femelles âgées de huit semaines, réparties aléatoirement en trois groupes : un groupe témoin négatif, un groupe modèle EAE et un groupe EAE traité par fingolimod. Le fingolimod a été administré dans l'eau de boisson à la dose de 0,5 mg/kg/jour à partir du 18<sup>e</sup> jour suivant l'induction de l'immunisation.
L'équipe de recherche a recueilli simultanément des données comportementales et moléculaires :Les indicateurs comportementaux couvrent la capacité motrice, la coordination physique et le comportement social ; au niveau moléculaire, la technologie de quantification ciblée LC-MS/MS a été utilisée pour détecter la concentration de 62 médiateurs lipidiques dans quatre tissus : le plasma, le cervelet, l'hippocampe et le cortex préfrontal, couvrant quatre grandes catégories : l'acide lysophosphatidique, les céramides, les sphingolipides et les endocannabinoïdes.Enfin, une matrice de données standard de « caractéristiques individuelles de la souris × lipides » a été construite.
Avant l'analyse des données,L'équipe de recherche a effectué une transformation logarithmique sur les données de concentration lipidique afin de les rendre conformes aux hypothèses de distribution de l'analyse statistique.Pour les valeurs manquantes dans les données originales 5.3%, après alignement multi-méthodes, ces valeurs ont été imputées à l'aide de l'algorithme Random Forest (missForest). Une analyse de variance à un facteur (ANOVA) a ensuite été réalisée sur 62 indicateurs lipidiques, et la correction de Šidák a été appliquée pour contrôler les erreurs de comparaisons multiples. Parallèlement, trois modèles d'apprentissage automatique – Random Forest, SVM et k-plus proches voisins – ont été utilisés pour valider la stabilité des signaux biologiques dans les données selon deux dimensions : la significativité des différences intergroupes et la capacité de prédiction de la classification.
Après avoir réalisé l'analyse de base, l'étude a mené une expérience de validation essentielle : la réduction systématique de la taille de l'échantillon afin de déterminer la valeur critique au-delà de laquelle les résultats statistiques sont erronés avec de petits échantillons. Les chercheurs ont progressivement réduit le nombre de souris dans chaque groupe, en répétant l'ensemble de la procédure d'analyse après chaque réduction. Les résultats ont montré que lorsque la taille de l'échantillon était réduite à 6 souris par groupe,La disparition complète de tous les résultats statistiques significatifs des données originales sert de référence pour évaluer les capacités d'augmentation des données de genESOM.—Dans les cas où les échantillons sont petits et où les statistiques sont totalement inefficaces, vérifiez si l'IA peut récupérer des signaux biologiques masqués par le bruit.
Intelligence artificielle générative conçue spécifiquement pour les données biomédicales issues de petits échantillons
Les modèles génératifs traditionnels sont confrontés à un dilemme récurrent lors du traitement de données issues de petits échantillons : soit les données générées manquent d’informations et ne permettent pas de retrouver les signaux biologiques originaux, soit elles sont surchargées de bruit, produisant un grand nombre de faux positifs. Le principe fondamental de genESOM est d’établir un mécanisme d’équilibre rigoureux entre ces deux facteurs, permettant ainsi une augmentation des données issues de petits échantillons sûre et interprétable.
genESOM est basé sur le réseau neuronal Emergent Self-Organizing Map (ESOM) et apporte deux améliorations clés par rapport à la carte auto-organisatrice (SOM) classique :Premièrement,Les neurones sont disposés dans une grille circulaire bidimensionnelle afin de préserver au maximum les relations de structure de voisinage des données de haute dimension.Deuxièmement,L'ajout d'une troisième dimension, codant l'espacement des sous-groupes et l'erreur de projection, améliore considérablement la précision de l'identification des structures de regroupement potentielles.
Après standardisation et suppression des valeurs manquantes, les données sont projetées sur le réseau ESOM pour l'entraînement. Le modèle sélectionne en continu le neurone optimal pour chaque échantillon, ajuste dynamiquement les poids des neurones et réduit progressivement le taux d'apprentissage afin d'assurer la stabilité de l'entraînement. À l'issue de l'entraînement, le modèle produit deux matrices principales : la matrice U, qui caractérise l'espacement entre les neurones et identifie les limites des clusters ; et la matrice P, utilisée pour analyser statistiquement la densité locale des données et servir de base à la génération de données synthétiques. Le paramètre de rayon, qui contrôle la portée de la génération des données synthétiques, est déterminé automatiquement par l'ajustement d'une distribution de distance à l'aide d'un modèle de mélange gaussien, sans intervention manuelle.
La conception la plus novatrice de genESOM réside dans la séparation complète des processus d'apprentissage de la structure et de génération de données.Le modèle apprend d'abord indépendamment la représentation de la structure inhérente des données, puis génère des données synthétiques à partir de cette structure stable, évitant ainsi l'accumulation d'erreurs lors des deux étapes. Plus important encore, le modèle peut introduire des variables de permutation comme contrôles négatifs afin de surveiller en temps réel si l'importance des caractéristiques est anormalement amplifiée ; dès qu'une accumulation d'erreurs est détectée, l'augmentation des données est immédiatement et automatiquement interrompue, réduisant ainsi les risques de surapprentissage et de faux positifs.
Dans cette étude, l'équipe de recherche a utilisé un ratio d'amélioration sûr de 1:1 (un échantillon synthétique généré à partir de chaque échantillon original) pour augmenter la taille de l'échantillon dans chaque groupe de 6 à 12. Après amélioration,Un ensemble complet d'analyses statistiques et d'apprentissage automatique est réalisé sur les données originales afin d'évaluer quantitativement l'effet de récupération du signal.Parallèlement, l'étude a comparé genESOM directement à deux méthodes génératives courantes : le modèle de mélange gaussien (GMM) et le réseau antagoniste génératif à table conditionnelle (CT-GAN), en utilisant le taux de faux positifs, le taux de faux négatifs et le taux de récupération du signal original comme indicateurs principaux pour vérifier les avantages du modèle.
Elle surpasse nettement les méthodes de génération traditionnelles dans les scénarios avec de petits échantillons.
Comme le montre la figure ci-dessous, l'analyse de l'ensemble des données initiales révèle des différences intergroupes significatives pour 27 des 62 variables lipidiques, les variations des lysophosphatidylcholines étant les plus significatives. Ce résultat est tout à fait cohérent avec les conclusions de recherches antérieures sur la sclérose en plaques. Parallèlement, le modèle de forêt aléatoire classe les échantillons avec une précision bien supérieure à celle obtenue par une classification aléatoire, corroborant ainsi ces deux résultats.Ceci confirme la présence de signaux biologiques stables et fiables dans les données originales.
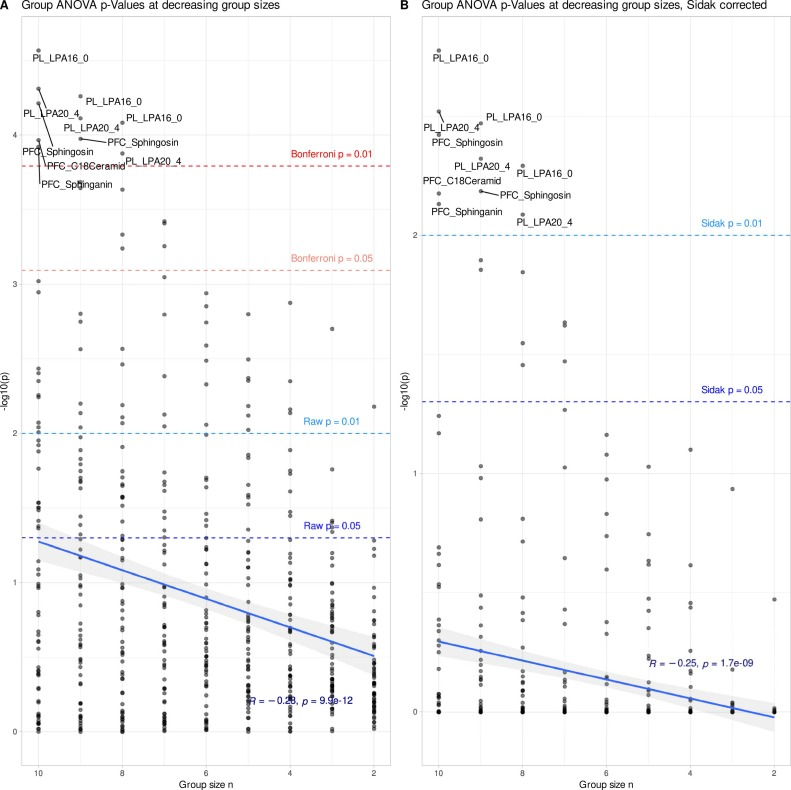
Cependant, lorsque la taille de l'échantillon dans chaque groupe a été réduite à 6 animaux, comme illustré dans la figure ci-dessous, les caractéristiques des données ont radicalement changé : après plusieurs corrections de validation, la significativité statistique de tous les indicateurs lipidiques a complètement disparu, et l'efficacité de classification de la forêt aléatoire a également diminué de manière significative. Il est important de souligner que cela ne signifie pas que les effets biologiques ont véritablement disparu.En revanche, la petite taille de l'échantillon entraîne une puissance de détection statistique insuffisante, et le véritable signal est noyé sous le bruit.
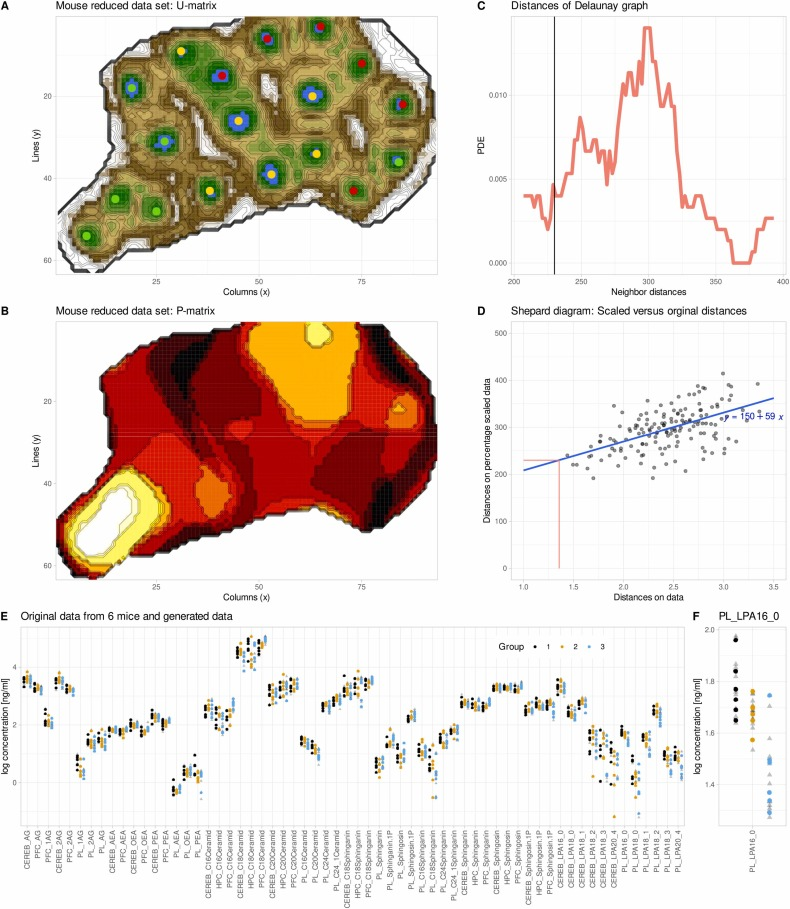
Par la suite, l'équipe de recherche a utilisé genESOM pour compléter les données réduites.Après 20 cycles d'entraînement, le modèle était encore capable d'identifier certaines tendances de séparation des trois groupes d'échantillons dans l'espace ESOM.Cela confirme que même lorsque la signification statistique disparaît, les données conservent encore des informations structurelles biologiques potentielles.
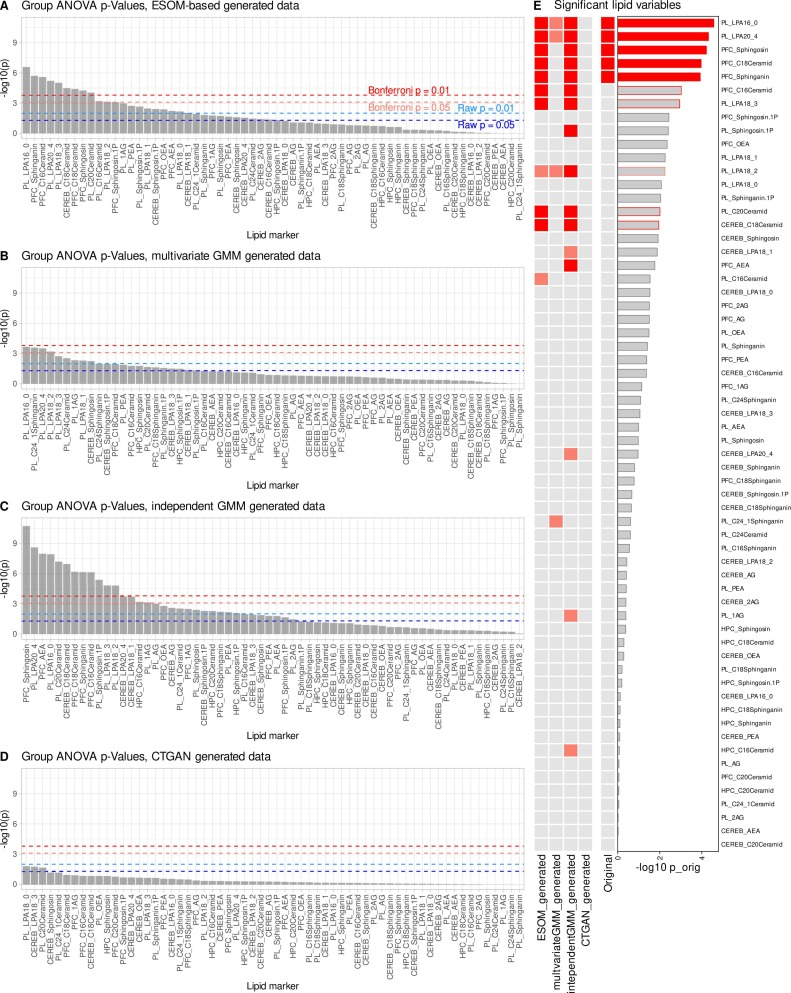
Après augmentation des données, des indicateurs lipidiques clés, tels que l'acide lysophosphatidique et les sphingolipides plasmatiques du cortex préfrontal, ont de nouveau présenté des différences intergroupes significatives. Ces indicateurs, totalement inopérants sur un petit échantillon, ont été rétablis avec succès grâce à l'augmentation des données par intelligence artificielle. Parallèlement, le modèle n'a pas introduit un grand nombre de nouvelles caractéristiques non pertinentes, et seuls quelques indicateurs supplémentaires, présentant des niveaux de signification proches, sont apparus.Cela indique que genESOM ne crée pas de nouveaux signaux à partir de rien, mais amplifie plutôt de véritables signaux biologiques qui existent déjà mais qui ne peuvent pas être détectés en raison d'une taille d'échantillon insuffisante.
Dans les mêmes conditions de petit échantillon, comme illustré dans la figure ci-dessous, les deux méthodes de génération de contrôle ont donné de mauvais résultats : le modèle de mélange gaussien multivarié n’a pu récupérer qu’une partie du signal original ; le modèle de mélange gaussien indépendant, bien qu’ayant récupéré certains indicateurs significatifs, a présenté un nombre important de faux positifs ; et le GAN à table conditionnelle n’a pas permis de récupérer efficacement les résultats principaux, avec un taux de faux négatifs élevé. Globalement,genESOM démontre une stabilité et une fiabilité nettement supérieures aux méthodes de génération traditionnelles dans les scénarios à petits échantillons.Il peut récupérer avec précision les signaux biologiques clés tout en contrôlant strictement la propagation des erreurs et les fausses découvertes.
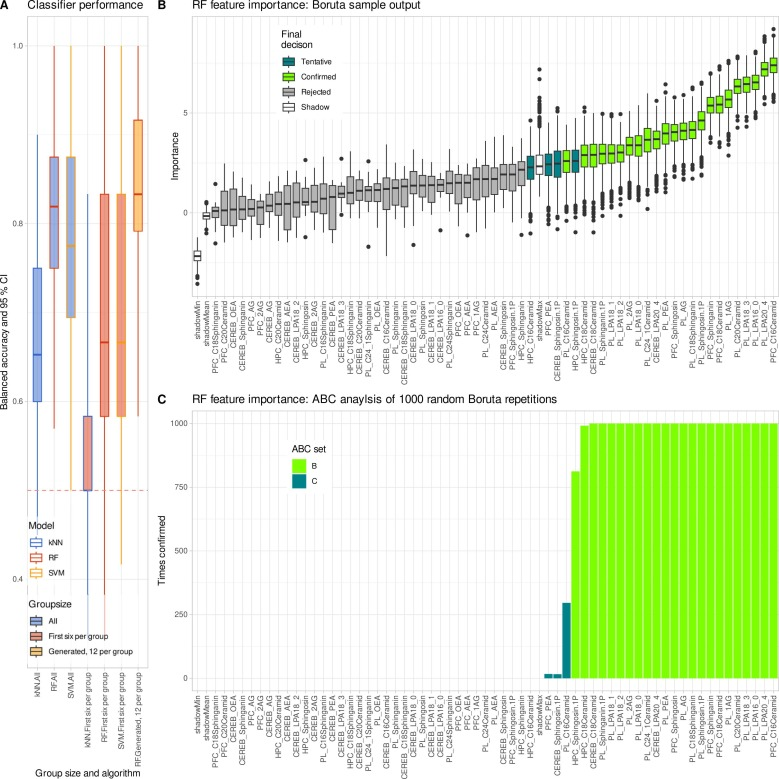
Comme le montre la figure ci-dessous, l'analyse d'apprentissage automatique a confirmé cette conclusion : les données améliorées ont rétabli la capacité de classification de la forêt aléatoire, et les caractéristiques clés sélectionnées étaient très cohérentes avec l'étude originale.
Derniers mots
La petite taille des échantillons constitue depuis longtemps un défi majeur en recherche biomédicale : coûts élevés, obstacles éthiques et difficultés d’obtention d’échantillons entraînent une puissance statistique insuffisante. L’augmentation de données traditionnelle est limitée par des problèmes de comparabilité, et l’IA générative générale est sujette aux faux positifs avec de petits échantillons. L’innovation de genESOM réside non pas dans la « fabrication » de données, mais dans la récupération progressive de signaux biologiques existants à partir de données limitées.
Sa conception fondamentale dissocie l'apprentissage de la structure de la génération de données, corrige les erreurs par ajustement de la dimensionnalité et introduit un contrôle négatif pour la surveillance en temps réel, formant ainsi un cadre rigoureux visant à « améliorer l'existant sans créer l'inexistant ». Il est important de noter que cette amélioration ne saurait remplacer les expérimentations réelles ; cette méthode est encore au stade exploratoire et son applicabilité nécessite d'être validée. Toutefois, cette recherche envoie un signal fort : sous un contrôle strict des erreurs et des faux positifs, l'IA générative a le potentiel de devenir un outil auxiliaire efficace pour les études sur petits échantillons, contribuant à dégager plus fiablement des conclusions pertinentes à partir de données limitées.








