Command Palette
Search for a command to run...
Une Centaine d'universités Ont Lancé La Plus Grande Étude Protéogénomique Multicohorte Au Monde, Permettant d'identifier Des Gènes Responsables De Maladies Et De Réorienter l'utilisation De Médicaments Existants Grâce Aux Données De Près De 80 000 participants.

Le génome humain est comme un mode d'emploi complet pour la vie, enregistrant toutes les informations génétiques telles que l'apparence, la taille, la morphologie et les prédispositions aux maladies. Cependant, déchiffrer ce manuel n'est pas chose simple ; divers événements imprévus peuvent survenir, notamment des mutations pathogènes qui prédisposent les individus à certaines maladies. Plus complexe encore…La plupart des variants pathogènes sont situés dans des régions non codantes du génome qui ne codent pas directement de protéines.Ce mécanisme de « boîte noire », qui ne précise ni quel gène est responsable de la maladie ni par quel mécanisme, limite considérablement notre capacité à identifier les gènes et les mécanismes pathogènes. Et en tant qu'exécuteurs directs qui donnent vie aux fonctions des gènes,Les milliers de protéines circulant dans le sang humain sont essentielles pour décrypter les mécanismes opaques et relier les variations non codantes aux mécanismes liés aux maladies.
Actuellement, la recherche en protéogénomique a réalisé des progrès significatifs dans la compréhension de la pathogenèse clinique et l'identification de cibles thérapeutiques potentielles, mais son application systématique et à grande échelle en biologie humaine reste limitée. Premièrement, les recherches antérieures se sont presque exclusivement concentrées sur les variants cis-régulateurs proximaux (c.-à-d. les loci de caractères quantitatifs des protéines cis, cis-pQTL).Les variations non codantes peuvent se situer dans des régions régulatrices, affectant ainsi directement plusieurs gènes codants voisins.Il peut également réguler indirectement et à distance les protéines codées par des gènes situés à d'autres endroits du génome ; deuxièmement, les recherches antérieures sur la structure génétique multigénique des biomarqueurs protéiques affectant le diagnostic et le pronostic des maladies restent insuffisantes ; enfin, pour identifier de manière stable et générale les loci de caractères quantitatifs protéiques, une validation répétée dans différentes populations est nécessaire.Actuellement, très peu d'études de validation humaine de ce type sont menées dans le domaine de la protéomique à large spectre.
Compte tenu de cela,Une équipe composée de chercheurs de plus d'une centaine d'universités et d'instituts de recherche, dont l'université Queen Mary de Londres et l'université de Cambridge, a publié la plus vaste étude protéogénomique multicohorte au monde à ce jour.Sur la base d'une méta-analyse à grande échelle des génomes protéoglycémiques couvrant 38 cohortes de recherche indépendantes et un total de 78 664 sujets, 24 738 sites de traits quantitatifs de protéines ont été systématiquement identifiés et associés à 1 116 protéines circulantes, révélant de manière exhaustive les caractéristiques de régulation génétique de proximité et de distance étendues au niveau des protéines.
L'apprentissage automatique a permis d'analyser plus en détail les voies de signalisation clés, les types cellulaires et les tissus d'origine régulant l'abondance des protéines circulantes, clarifiant ainsi le rôle central de la N-glycosylation dans le réseau de régulation protéique. De plus, la distinction entre les différences de régulation cis et trans au sein des protéines permet d'élucider efficacement les mécanismes intrinsèques de différents phénotypes biologiques, fournissant des éléments pour le criblage de cibles protéiques potentielles pour le développement de médicaments dans certaines maladies. Enfin, l'analyse par triangulation des sites trans a révélé des preuves plus solides en faveur du repositionnement de médicaments.
Les résultats de cette recherche, intitulée « Des analyses protéogénomiques multicohortes révèlent des effets génétiques à travers le protéome et le diséasome », ont été publiés dans la revue Cell.
Points saillants de la recherche :
* La plus grande étude protéogénomique multicohorte à ce jour, englobant 38 cohortes d'études indépendantes et impliquant un total de 78 664 participants.
* 24 738 loci de caractères quantitatifs de protéines ont été identifiés et associés à 1 116 protéines circulantes, révélant de manière exhaustive un large éventail de caractéristiques de régulation génétique de proximité et à longue distance au niveau des protéines.
Cette étude élucide systématiquement les mécanismes de régulation des protéines circulantes au niveau génétique, fournissant une base théorique et des ressources de données importantes pour comprendre les mécanismes moléculaires des maladies humaines, identifier des cibles thérapeutiques innovantes et mener des recherches sur le repositionnement de médicaments.

Adresse du document :
https://www.cell.com/cell/fulltext/S0092-8674(26)00385-5
Données de base à grande échelle : 38 cohortes internationales, près de 80 000 participants
Cette étude représente la plus grande méta-analyse protéogénomique multicohorte au monde.Intégrant 38 cohortes internationales, couvrant 78 664 participants d'origine européenne,Sur la base de l'analyse de 1 161 cibles protéiques sanguines à l'aide de la technologie protéomique à haut débit Olink, 24 738 pQTL finement cartographiés (dont 5 040 cis-pQTL et 19 698 trans-pQTL) ont été identifiés, et des données de régulation génétique pour 1 116 protéines efficaces ont été obtenues.
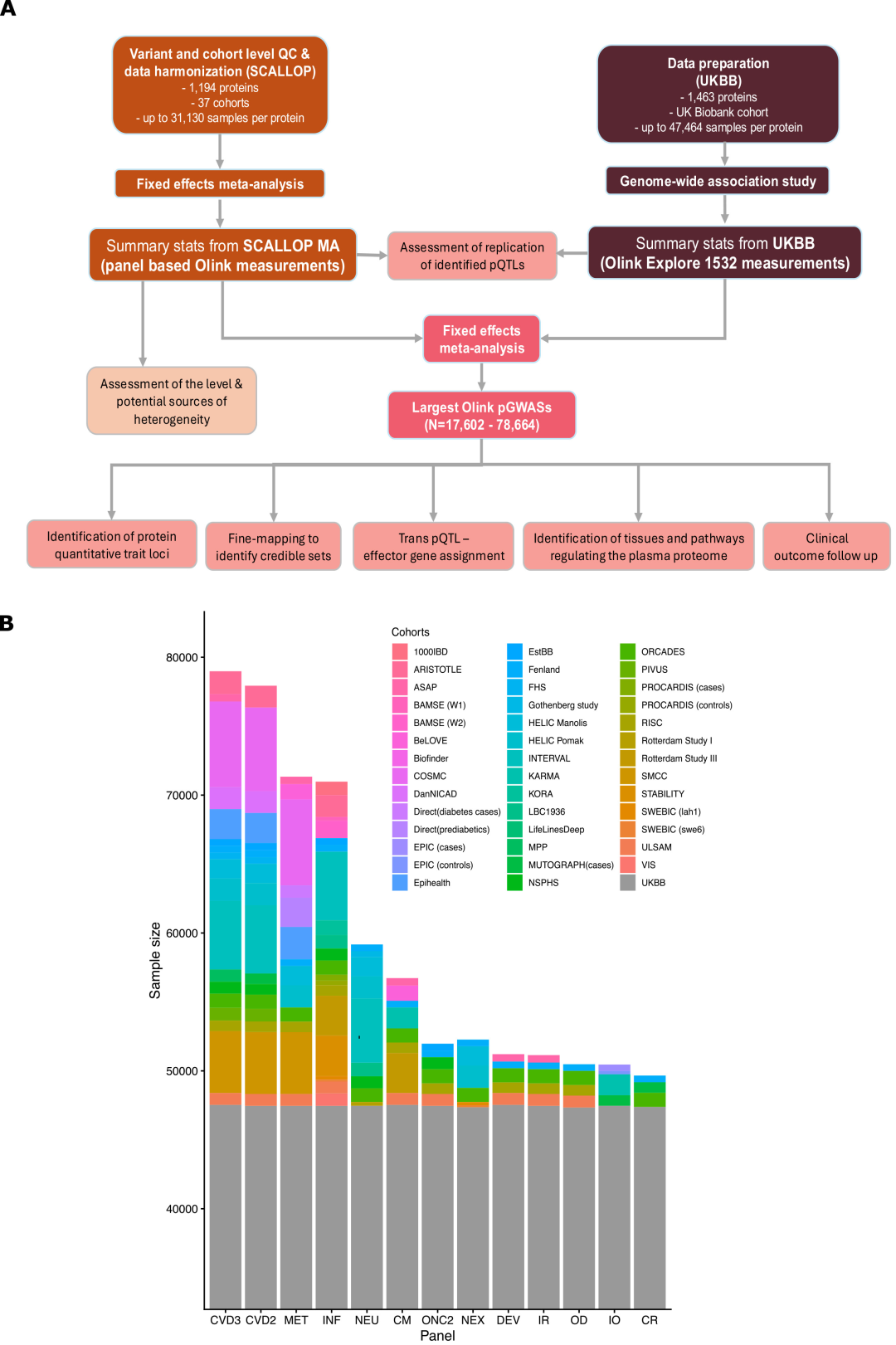
Méta-analyses sur la coquille Saint-Jacques :Ces données comprennent des statistiques génomiques complètes issues de 37 cohortes et portant sur 1 194 cibles protéiques sanguines, la majorité des participants étant d’origine européenne. Des analyses protéomiques basées sur des anticorps ont été réalisées à l’aide d’au moins un des 13 panels d’analyse Target-96 fournis par Olink, chacun capable de détecter 92 cibles protéiques couvrant les domaines cardiovasculaire, immunitaire, inflammatoire, neurologique et métabolique.
Biobanque du Royaume-Uni (UKBB) :L'étude a porté sur 48 017 participants d'origine européenne. Les données protéomiques utilisées ont été générées à l'aide de la plateforme Olink Explore 1536, qui a également employé des techniques basées sur des anticorps pour mesurer 1 463 cibles protéiques.
Classificateur d'apprentissage automatique par étapes
L'objectif principal de cette étude, qui utilise des modèles d'apprentissage automatique, est d'attribuer systématiquement, précisément et à grande échelle des « gènes effecteurs » à tous les trans-pQTL situés en dehors de la région du complexe majeur d'histocompatibilité (CMH). Ceci permet de relever le défi de longue date que représente la localisation des gènes effecteurs dans des régions génomiques éloignées des loci de caractères quantitatifs protéiques associés aux taux de protéines sanguines. À cette fin,S'inspirant de l'architecture ProGeM, des chercheurs ont construit un classificateur d'apprentissage automatique par étapes.
Premièrement, concernant les sources des caractéristiques et des annotations, les chercheurs ont intégré des annotations biologiques et génomiques multidimensionnelles pour chaque variant génétique ou ses variants alternatifs (r² > 0,6). Les annotations au niveau du variant incluaient la distance entre le variant et le génome dans une fenêtre de 1 Mb et l'impact fonctionnel potentiel déduit à l'aide de l'outil de prédiction de l'effet du variant (VEP).
Simultanément, une annotation au niveau du gène a été réalisée pour chaque gène dans une fenêtre de base de 1 Mb, incluant l'obtention de preuves pertinentes de colocalisation QTL basées sur l'abondance des protéines GTEx v8 et l'expression génique, l'association de charge de variants rares, l'utilisation du package OmnipathR version 3.10.1 pour examiner la littérature et déterminer s'il existe des complexes ligand-récepteur/protéine correspondant aux protéines cis codées par les gènes trans, et la détermination de la participation des gènes apparentés à la même voie biologique sur la base des informations d'annotation KEGG/REACTOME.
Ensuite, nous procédons à la constitution de l'ensemble d'entraînement nécessaire au modèle d'apprentissage automatique, en raison du manque de variantes de référence largement utilisées pour l'attribution des gènes.Les chercheurs ont utilisé leurs connaissances préalables en biologie et en génomique pour obtenir trois ensembles partiellement indépendants de « vrais positifs présumés (VPP) ».Afin d'éviter tout biais, une seule protéine cis a été conservée au sein de chaque ensemble PTP, les autres gènes situés dans une fenêtre de 1 Mb étant considérés comme des échantillons négatifs. Plus précisément, cela incluait les gènes trans codant pour des paires ligand-récepteur ou formant des complexes protéiques à haute confiance avec des protéines cis (n = 540), les trans-pQTL sentinelles associés à des variants fonctionnels (n = 1747) et les gènes trans présentant des charges de variants rares significatives (n = 1049). Les ensembles d'entraînement et de test ont ensuite été divisés selon un ratio de 7:3 en fonction des régions génomiques, et les résultats ont été reproduits 10 fois afin d'en garantir la stabilité.
Par ailleurs, concernant l'architecture du modèle et le processus d'apprentissage, l'algorithme utilisé dans cette étude est un classificateur de type forêt aléatoire. Dix ensembles d'apprentissage sont utilisés, et une validation croisée à trois plis est effectuée de manière répétée, combinée à une stratégie de sous-échantillonnage, afin de gérer le problème des ensembles de données déséquilibrés lors de l'apprentissage.L'entraînement du modèle a été mis en œuvre à l'aide de la boîte à outils caret v6.0.94 du langage R, puis le modèle de forêt aléatoire le plus performant dans chaque ensemble d'entraînement a été sélectionné grâce à l'évaluation du score Kappa.
Ensuite, à l'aide de 10 classificateurs de forêt aléatoire correspondant à chaque jeu de données hypothétique de vrais positifs, tous les gènes effecteurs candidats de trans-pQTL ont été évalués individuellement. Le score médian des 10 classificateurs pour chaque jeu de données hypothétique de vrais positifs a d'abord été calculé, puis les trois ensembles de scores prédits ont été additionnés. Simultanément, lors de la construction du modèle de classification pour chaque jeu de données hypothétique de vrais positifs, les variables de caractéristiques utilisées pour définir les échantillons de vrais positifs ont été supprimées.
Au final, les trois modèles de classification ont démontré des performances stables et fiables, avec des coefficients Kappa médians allant de 0,54 à 0,57.
Le décryptage du mécanisme de la pathogenèse fournit des preuves génétiques pour le développement de médicaments et la réutilisation de médicaments existants.
Cette étude, basée sur 38 cohortes internationales et portant sur 78 664 participants, a mené une méta-analyse génomique des protéines multicohortes ciblant 1 161 cibles protéiques sanguines, élucidant systématiquement les modèles de régulation génétique des niveaux de protéines circulantes et leur association avec la maladie.
Identification et caractéristiques des pQTL
L'étude a identifié 14 690 variants sentinelles régionaux, et le mappage fin bayésien a permis d'obtenir 24 738 ensembles de variants indépendants et fiables, comprenant 5 040 cis-pQTL et 19 698 trans-pQTL, couvrant 1 116 cibles protéiques. Parmi ceux-ci, des cis-pQTL étaient présents dans la protéine située à la position 87.1%, et des trans-pQTL dans celle située à la position 94.1% ; les cis-pQTL situés à la position 82.3% et les trans-pQTL situés à la position 83.3% étaient des sites à haute confiance, contenant 278 cis-pQTL et 4 013 trans-pQTL nouvellement découverts. De plus, dans la cohorte d'ascendance non européenne, les tailles d'effet des sites identifiés ont montré une corrélation modérée avec celles de la cohorte européenne (r = 0,6).Cela confirme la robustesse des résultats pour différentes populations.
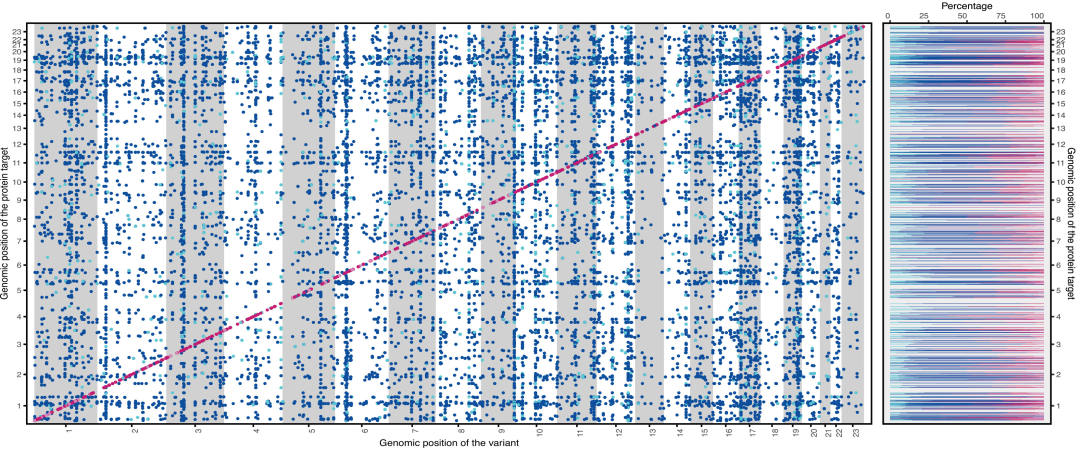
Localisation précise des loci de caractères quantitatifs protéiques dans les méta-analyses SCALLOP et UKBB
De plus, le pouvoir explicatif des loci génétiques pour les variations des taux de protéines sanguines présente des différences significatives. Les pQTL cis expliquent en moyenne 8,41 TP3T de la variation protéique, soit un pourcentage significativement plus élevé que celui des pQTL trans. Cependant, des protéines telles que ICAM2 et FUCA1 sont principalement régulées par des pQTL trans, avec des pouvoirs explicatifs respectifs de 52,71 TP3T et 68,41 TP3T, tandis que les pQTL cis n'expliquent que 0,31 TP3T et 6,31 TP3T.
De plus, l'observation plus poussée de 261 cibles protéiques n'a révélé aucune corrélation linéaire significative entre le pouvoir explicatif de leurs variations pQTL et l'héritabilité polygénique, ce qui suggère que cette étude a peut-être atteint une quasi-saturation dans l'identification des pQTL pour ces protéines.
Caractéristiques des protéines cibles sous régulation génique
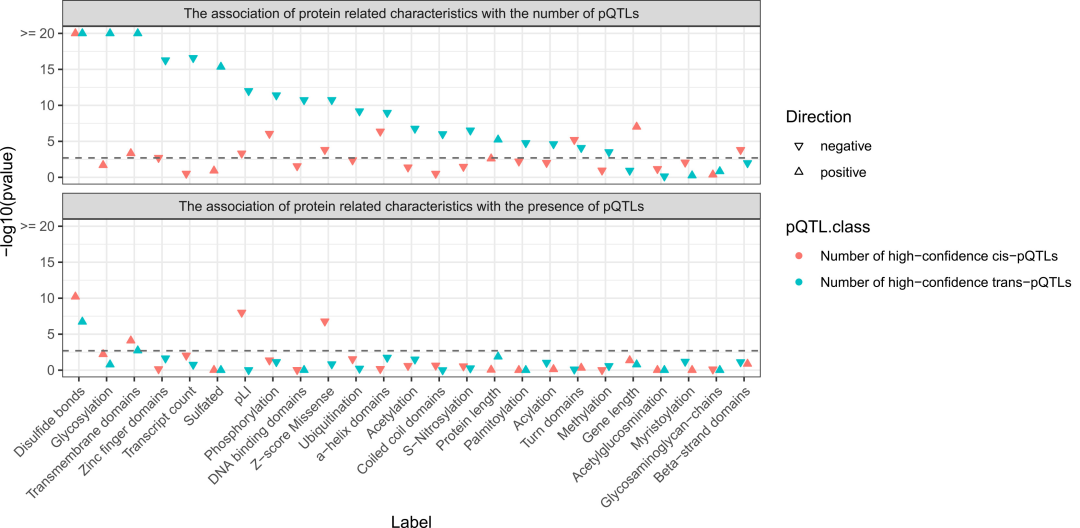
Caractéristiques protéiques liées à la présence et à la quantité de pQTL, basées sur un modèle de régression de Poisson à inflation de zéros.
Les protéines contenant des ponts disulfures et des domaines transmembranaires possèdent un nombre significativement plus élevé de pQTL, ce qui peut expliquer pourquoi ces protéines sont plus facilement régulées génétiquement ; tandis que la force de contrainte fonctionnelle des gènes codant pour les protéines est significativement corrélée négativement avec le nombre de cis-pQTL.
Les protéines présentant un nombre élevé de trans-pQTL sont significativement enrichies en caractéristiques de protéines sécrétoires telles que la glycosylation et la sulfatation, mais sont dépourvues de caractéristiques de protéines intracellulaires telles que les structures à doigts de zinc et les domaines de liaison à l'ADN, ce qui indique que la régulation génétique à longue distance des protéines circulantes est étroitement liée à la voie sécrétoire.
Analyse des gènes effecteurs trans-pQTL et des voies de régulation
Sur la base de l'intégration de connaissances biologiques antérieures dans le cadre d'apprentissage automatique, au moins un gène effecteur avec une confiance modérée a été identifié pour plus de la moitié des trans-pQTL (n = 11 261), dont 1 534 étaient des attributions à haute confiance ; pour les deux tiers des sites (n = 13 881), la distribution des scores candidats à travers les gènes a indiqué qu'un seul gène causal était le gène pathogène le plus probable.
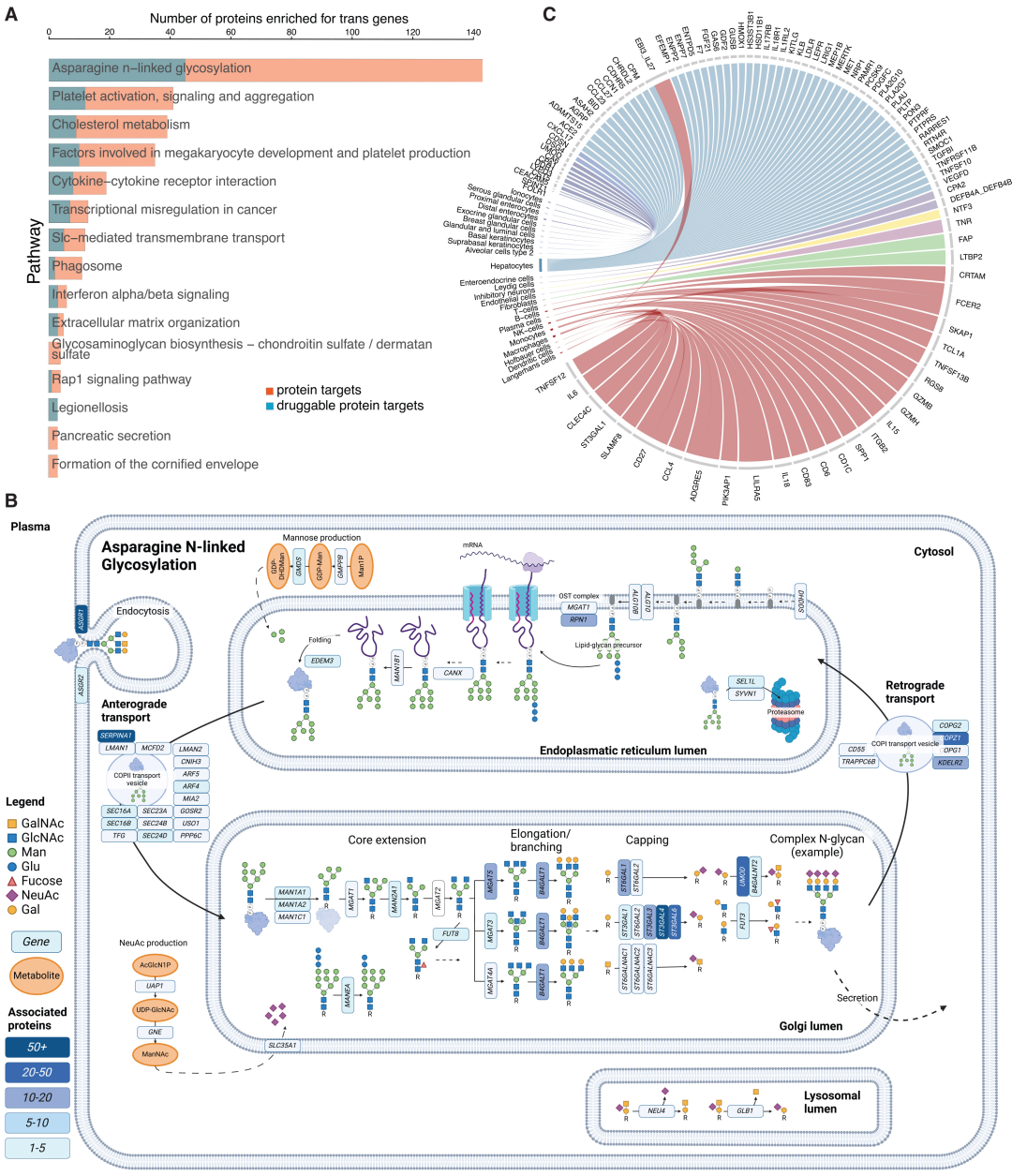
L'analyse d'enrichissement fonctionnel a montré que les gènes à effet trans étaient significativement enrichis dans la voie de N-glycosylation de l'asparagine (impliquant 143 cibles protéiques) et l'activation plaquettaire (impliquant 41 cibles protéiques), entre autres.La N-glycosylation est la voie de régulation la plus courante et la plus importante.
Les résultats d'enrichissement cellulaire et tissulaire ont montré que les gènes à effet trans étaient fortement exprimés principalement dans les hépatocytes, les cellules NK, les cellules endothéliales et les pneumocytes de type II, révélant que le foie et les cellules immunitaires sont des sites clés pour la régulation à distance des protéines circulantes. 44 paires protéine-tissu et 76 paires protéine-type cellulaire étaient d'origine sécrétoire non classique, confirmant le rôle important de la communication inter-organes dans la régulation de l'homéostasie protéique.
Effets pléiotropiques aux niveaux moléculaire et phénotypique
Parmi tous les pQTL indépendants identifiés, 43,41 TP3T ont présenté des effets pléiotropiques, les pQTL trans montrant des effets pléiotropiques significativement plus élevés que les pQTL cis. Des études ultérieures ont classé les variations génétiques pléiotropiques en trois types : « pléiotropique moléculaire », « pléiotropique phénotypique » et « pléiotropique non spécifique ». Plus de la moitié (332 sur 533) ont présenté des effets pléiotropiques phénotypiques.En particulier, son expression a été multipliée par deux dans les hépatocytes, et elle a régulé préférentiellement la protéine cible par le biais de complexes protéiques, d'interactions ligand-récepteur et d'une synergie de voie.
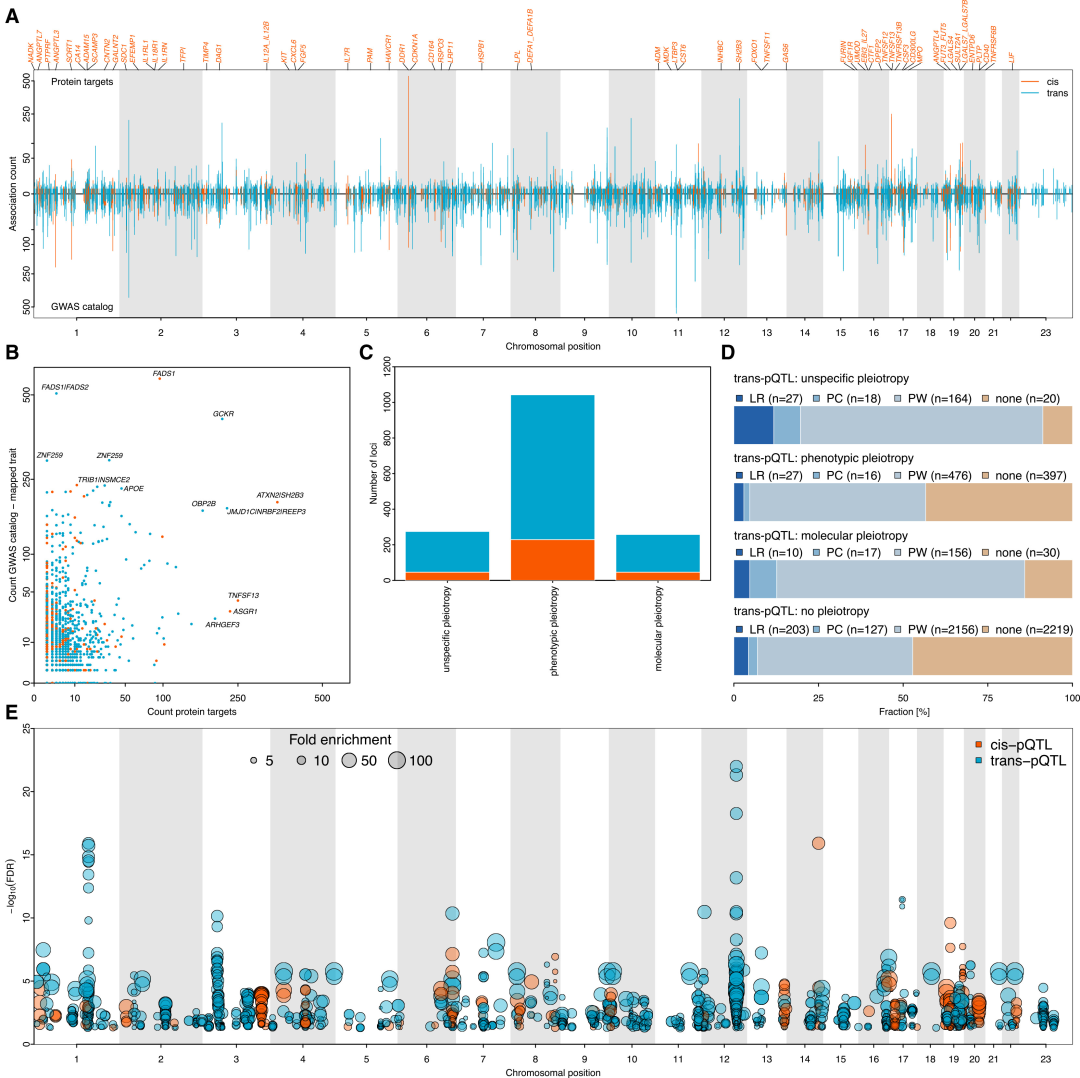
285 pQTL pléiotropiques chevauchent les sites GWAS de maladies, et leurs protéines associées sont significativement enrichies dans des voies spécifiques, fournissant de nouveaux indices pour élucider les mécanismes des sites GWAS de maladies.
Différences dans les phénotypes de la maladie sous régulation cis et trans
Des chercheurs ont combiné 300 associations protéine-maladie identifiées comme étant induites par des QTL cis-pQTL avec plus de 700 jeux de données de maladies provenant du projet FinnGen. Seules 73 de ces associations ont donné lieu à des analyses de colocalisation statistique qui ont permis de mettre en évidence à la fois la randomisation mendélienne et les signaux de risque génétique.Cela suggère que des preuves complémentaires sont nécessaires pour hiérarchiser les gènes potentiellement responsables de maladies.
Parmi les 115 associations évaluables, 31 ont montré des effets cis- et trans-régulateurs cohérents, 41 n'avaient aucune preuve à l'appui et 14 ont montré des effets opposés, indiquant une différence significative dans l'impact de la régulation cis-proximale et de la régulation trans-distale sur le phénotype de la maladie.
Analyse de l'association protéine-maladie dans les études d'inférence génétique et observationnelles
Cette étude intègre des données observationnelles issues de 52 164 participants à l’étude UKBB et des données génétiques provenant de plus de 1,29 million d’individus de la base de données PanBio, couvrant 517 maladies. Parmi les 193 associations génétiques à haute confiance, seules 52 ont été systématiquement confirmées par des études observationnelles ; et parmi les 52 887 associations observationnelles significatives, seulement 0,061 ont reçu une confirmation génétique. Notamment, la protéine furine sanguine est l’une des rares cibles systématiquement associées à l’hypertension, à l’infarctus du myocarde et à la fibrillation auriculaire dans les études génétiques et observationnelles, révélant son intérêt potentiel pour le développement de médicaments.
Les trans-pQTL guident la découverte de biomarqueurs de maladies et le reciblage de médicaments
Plus de 901 biomarqueurs protéiques de maladies TP3T (280 sur 307 maladies) étaient significativement enrichis en protéines associées à des trans-pQTL, confirmant que la transrégulation constitue le fondement génétique des biomarqueurs protéiques de maladies. L'étude a révélé que la mutation faux-sens rs34536443 du gène TYK2, en tant que trans-pQTL, régule plusieurs protéines inflammatoires telles que BST2 et CXCL9/10/11. Des niveaux élevés de ces protéines sont associés à un risque accru de polyarthrite rhumatoïde, de psoriasis et de thyroïdite auto-immune, fournissant ainsi des preuves génétiques pour le repositionnement des inhibiteurs de TYK2 dans le traitement des maladies auto-immunes.
Conclusion
Cette étude, fondée sur la plus vaste analyse protéogénomique multicohorte au monde, a élucidé de manière systématique les mécanismes de régulation génétique du protéome circulant humain. Elle a surmonté les limites des études précédentes qui se concentraient uniquement sur la régulation cis et a révélé pour la première fois de façon exhaustive le rôle clé de la régulation transgénique dans la régulation de l'abondance des protéines circulantes à grande échelle. De plus, elle a utilisé l'apprentissage automatique pour localiser précisément les gènes effecteurs, clarifiant ainsi des voies essentielles telles que la N-glycosylation et la biologie plaquettaire, ainsi que des sites de régulation clés comme le foie et les cellules immunitaires.
Bien que cette étude présente certaines limites, notamment le fait que la protéomique ne couvre que certains sous-types et modifications post-traductionnelles des protéines circulantes, et que la population étudiée soit principalement d'origine européenne (il serait nécessaire de l'étendre à d'autres groupes ethniques), elle établit néanmoins un cadre complet reliant les variations génétiques non codantes, les protéines circulantes et les mécanismes pathologiques. Elle offre non seulement une nouvelle perspective pour élucider les mécanismes moléculaires des maladies complexes, mais elle ancre également des cibles clés telles que la furine plasmatique et TYK2 grâce à des preuves génétiques. Ces preuves fournissent des éléments génétiques hautement crédibles pour le développement et le repositionnement de médicaments innovants, et constituent une étape cruciale dans la translation de la protéogénomique de la recherche fondamentale à l'application clinique.