Command Palette
Search for a command to run...
Débruitage Des données/amélioration Du Signal biologique/atténuation Des Pertes De Signal, Modèle d'apprentissage Profond SUICA Permet De Prédire l'expression Des Gènes À n'importe Quelle Position Dans Les Tranches De Transcriptome Spatial

Le groupe du professeur Zheng Yinqiang de l'Université de Tokyo et celui du professeur Ding Jun de l'Université McGill ont proposé conjointement une méthode de modélisation des données de transcriptome spatial, SUICA. SUICA est un modèle d'apprentissage profond basé sur des représentations neuronales implicites (INR) et des autoencodeurs de graphes. SUICA utilise des autoencodeurs de graphes pour réduire la dimensionnalité des données de transcriptome spatial de grande dimension, puis utilise des représentations neuronales implicites pour modéliser les coordonnées des données de transcriptome spatial et leurs expressions génétiques correspondantes, permettant ainsi de prédire l'expression génétique à n'importe quelle position dans la tranche de transcriptome spatial.Les résultats montrent que les données de transcriptome spatial traitées par SUICA peuvent avoir une meilleure qualité, un bruit plus faible et des signaux biologiques plus forts.
Les résultats connexes ont été sélectionnés pour ICML 2025 sous le titre « SUICA : Apprentissage de représentations neuronales implicites clairsemées de très haute dimension pour la transcriptomique spatiale ».

Adresse du document :
https://go.hyper.ai/C6Zcl
se concentrer sur 「HyperIA "Compte officiel WeChat, répondez « SUICA » dans les coulisses pour obtenir le PDF complet
Autres articles sur les frontières de l'IA :
https://go.hyper.ai/owxf6
Que sont les données du transcriptome spatial ?
Les données de transcriptomique spatiale (ST) sont une matrice d'informations de grande dimension qui enregistre simultanément les « niveaux d'expression génétique » et les « coordonnées spatiales » sur la même section de tissu.Par rapport à l'imagerie panoramique traditionnelle des tissus (WSI) qui ne peut présenter que des structures morphologiques ou à la transcriptomique conventionnelle qui ne peut que quantifier l'expression des gènes mais perd la direction, la transcriptomique spatiale lie « quels gènes sont exprimés » à « où ils sont situés dans le tissu », dessinant une carte fonctionnelle de l'interaction entre l'état cellulaire et le microenvironnement dans le tissu, devenant ainsi une nouvelle forme de données qui relie l'histologie et l'omique moléculaire.
Pourquoi est-il nécessaire d’améliorer les données transcriptomiques spatiales ?
Bien que la transcriptomique spatiale ait apporté des connaissances moléculaires résolues spatialement sans précédent, les données du monde réel sont toujours limitées par trois goulots d'étranglement majeurs :
1 Contradiction coût-résolution:Plus les sondes sont denses et plus la profondeur de séquençage est élevée, plus le coût expérimental (par exemple, le coût de l'expérience de séquençage du stéréo-seq est supérieur à $4 000/cm²) et le débit d'échantillon augmentent rapidement ;
2. Faiblesse du signal et bruit:Le nombre d'ARNm capturés à chaque point de détection est limité et l'expansion nulle est grave, ce qui facilite l'oubli de gènes régulateurs clés ou de faible abondance ;
③ Hétérogénéité multiplateforme:Différentes plates-formes présentent des différences significatives dans la disposition physique des sondes, la profondeur de séquençage et le bruit de fond, ce qui entrave directement l'intégration de plusieurs échantillons ou de plusieurs expériences.
Les méthodes d'amélioration informatique incluent la reconstruction en super-résolution, le débruitage profond et le remplissage des valeurs manquantes, qui peuvent faire ce qui suit sans augmenter (ou seulement légèrement) le coût expérimental :
(a) Prédire l’expression des gènes sur des sites qui n’ont pas été séquencés ;
(b) Récupérer la véritable expression génétique qui ne peut être détectée en raison de limitations techniques et améliorer la sensibilité de détection des gènes différentiellement exprimés et des gènes spatialement variables ;
(c) Générer des représentations de fonctionnalités standardisées qui sont comparables et partageables sur différentes plateformes.
Cela fournira une base de données plus précise, plus riche et plus évolutive pour l'analyse de la communication cellulaire, l'annotation du zonage des maladies, la découverte de cibles médicamenteuses, la modélisation conjointe multi-omique et le diagnostic assisté par la pathologie de l'IA, libérant ainsi considérablement le potentiel de la technologie transcriptomique spatiale dans la recherche fondamentale et la transformation clinique.
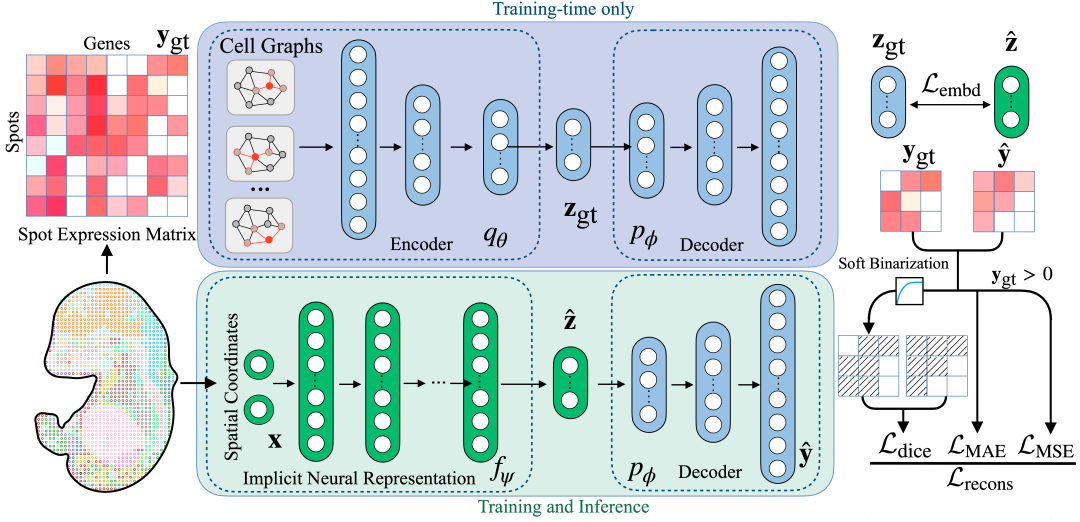
SUICA : Un modèle unifié basé sur une représentation neuronale implicite et un autoencodeur graphique
Défis de la modélisation des données transcriptomiques spatiales à l'aide de représentations neuronales implicites
La modélisation des données transcriptomiques spatiales est confrontée à de multiples défis :
Tout d’abord, les données originales sont distribuées sous forme de grille dans la dimension spatiale.En termes de dimension génétique, le nombre peut atteindre des milliers, voire des dizaines de milliers, formant une matrice « ultra-dimensionnelle, extrêmement clairsemée et bruyante » ; le taux d'abandon élevé affaiblit les signaux biologiques clés, exacerbant le manque de puissance statistique.
Deuxièmement, les plateformes de transcriptomique spatiale existantes présentent un compromis fondamental entre résolution et coût.——Plus les sondes sont denses et plus le séquençage est profond, plus le coût augmentera de manière exponentielle, ce qui rendra difficile d’atteindre simultanément une résolution au niveau cellulaire et des tailles d’échantillon à grande échelle.
Troisièmement, lorsqu'on essaie d'utiliser une représentation neuronale implicite pour interpoler des points discrets du transcriptome spatial dans des champs d'expression continus, deux difficultés techniques majeures doivent être résolues en même temps : premièrement, la dimension de l'espace d'expression génétique dépasse de loin celle des signaux visuels traditionnels, et le simple fait d'élargir ou d'approfondir le réseau est difficile à éliminer de la malédiction de la dimensionnalité ; deuxièmement, l'expansion nulle conduit à une distribution très inégale des signaux d'entrée, et les INR conventionnels sont difficiles à capturer des modèles d'expression spatiale complexes et non linéaires.
Figure Autoencoder : Réduction de la dimensionnalité des données du transcriptome dans un espace à haute dimension
Par rapport aux autoencodeurs traditionnels, nous considérons d'abord les points de données de chaque transcriptome spatial comme des nœuds de graphe et construisons une matrice d'adjacence basée sur la proximité spatiale. Ensuite, nous utilisons la convolution de graphe dans l'encodeur pour convoluer l'expression génétique originale de grande dimension, intégrer le contexte spatial local à la représentation et la compresser en une représentation de faible dimension. De cette manière, nous apprenons la représentation de faible dimension des données de transcriptome spatial de grande dimension, et l'ajout de la convolution de graphe peut améliorer le signal de données de transcriptome spatial clairsemé et bruité.
Représentation neuronale implicite : établissement d'une correspondance entre les coordonnées des points de séquençage et l'expression des gènes
Après avoir obtenu la représentation en basse dimension,Le réseau de représentation neuronale implicite reçoit les coordonnées des points de détection en entrée et apprend la correspondance entre le « point » et sa représentation de faible dimension correspondante.Et la représentation de faible dimension apprise et prédite par le modèle est envoyée à la partie décodeur de l'autoencodeur graphique, réalisant ainsi la fonction de mappage des coordonnées à l'expression génétique de haute dimension.
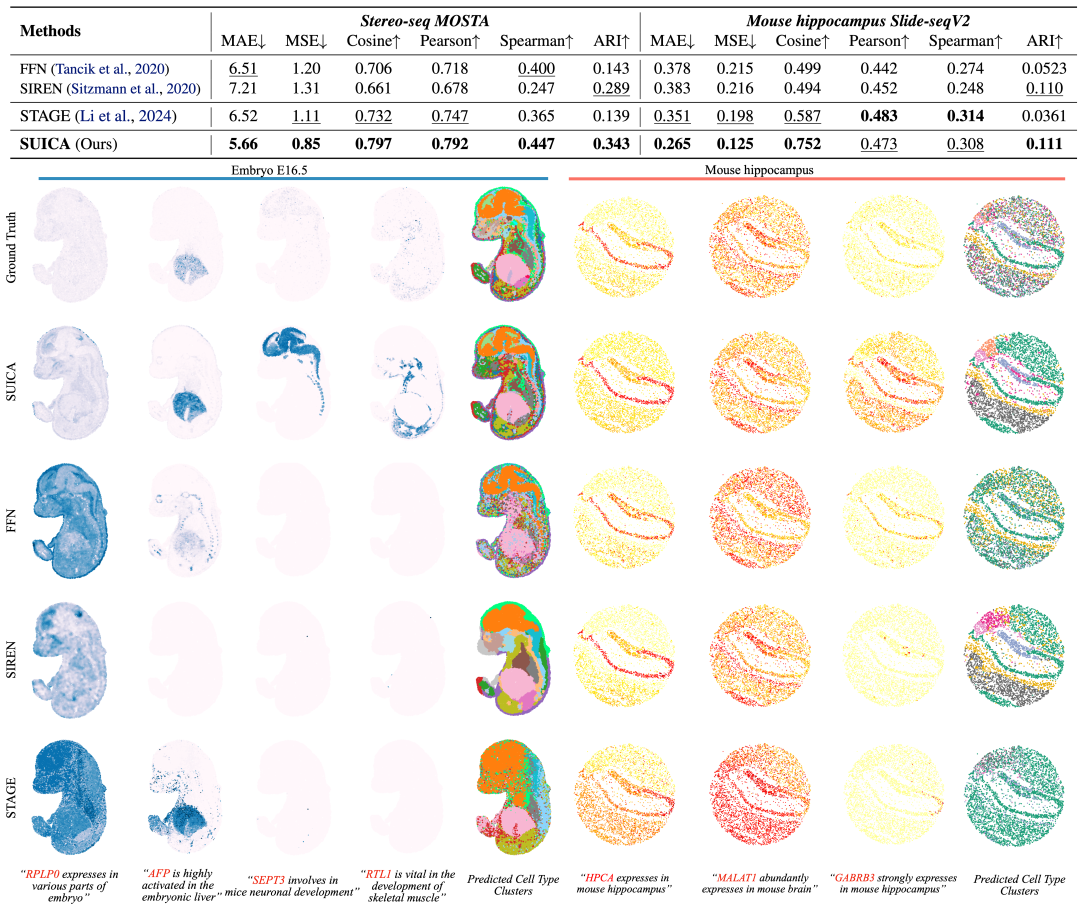
Vérification expérimentale : SUICA peut générer des résultats de prédiction plus précis et biologiquement pertinents
Nous avons utilisé des données d'embryons de souris stéréo-séquencées et des données de tranches de cerveau de souris par séquençage sur diapositives à des fins de comparaison. Dans la tâche de prédiction de point inconnu (super-résolution), SUICA a nettement surpassé les modèles existants et les modèles traditionnels de représentation neuronale implicite, notamment FFN et SIREN, sur plusieurs indicateurs clés. Nous avons visualisé l'effet prédictif de chaque méthode, et les résultats ont montré que la prédiction de SUICA permet non seulement de restaurer avec précision le profil d'expression des gènes, mais aussi d'en améliorer le signal. Par exemple, SEPT3, un gène jouant un rôle important dans le développement du système nerveux des embryons de souris, a réussi à capturer ce signal malgré le fait que le signal dans la vérité terrain ne soit pas évident.
En regroupant et en étiquetant les résultats obtenus par diverses méthodes, nous avons intuitivement constaté que les types cellulaires générés par SUICA sont les plus proches des types cellulaires réels. De plus, les types cellulaires générés par SUICA conservent des structures organiques et tissulaires plus détaillées dans l'espace.Ces résultats démontrent que SUICA a la capacité d’améliorer les signaux biologiques et d’identifier les différences subtiles dans les états cellulaires entre différents organes et tissus.
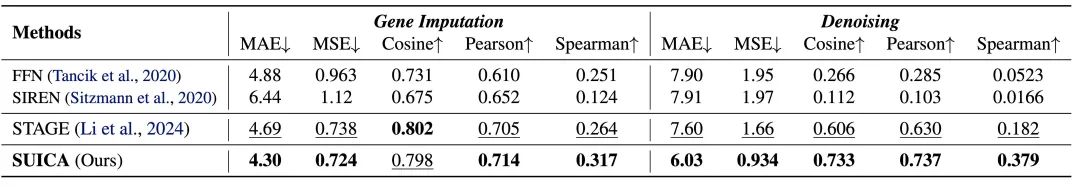
Vérification expérimentale : SUICA peut réduire le bruit des données du transcriptome spatial et atténuer le phénomène d'abandon
Pour vérifier la capacité de débruitage de SUICA (imputation génétique) et sa capacité à récupérer la véritable expression génétique à partir d'une perte (résultant de 0 lectures en raison des limitations de la technologie de séquençage), nous avons ajouté artificiellement du bruit gaussien aux données du transcriptome spatial ou défini aléatoirement l'expression génétique à 0. Dans l'expérience d'imputation génétique, nous avons défini aléatoirement 70 % de l'expression génétique dans les données à 0. Dans l'expérience de débruitage de l'expression génétique, afin de garantir que la distribution de l'expression génétique après l'ajout de bruit est toujours similaire à la distribution de l'expression génétique d'origine, nous avons défini toutes les valeurs négatives à zéro.Les résultats expérimentaux montrent que SUICA est supérieur aux méthodes existantes dans de multiples indicateurs, prouvant sa capacité à réduire le bruit dans les données du transcriptome spatial et à atténuer le phénomène d'abandon.