Command Palette
Search for a command to run...
DeepMind Lance AlphaGenome, Qui Prédit Les Effets Des Mutations Sur Toutes Les Modalités Et Tous Les Types De Cellules En 1 Seconde

La série Alpha de Google DeepMind ajoute un nouveau membre - AlphaGenome,Il peut prédire de manière plus complète et plus précise comment une seule variation ou mutation dans une séquence d’ADN humain affecte une série de processus biologiques qui régulent les gènes.
Le modèle AlphaGenome prend en entrée une séquence d'ADN pouvant contenir jusqu'à 1 million de paires de bases et prédit des milliers de propriétés moléculaires liées à son activité régulatrice.Parallèlement, l'impact de la variation ou de la mutation génétique peut être évalué en comparant les résultats de prédiction des séquences variantes et non variantes. Ce modèle s'appuie sur le modèle génomique Enformer de DeepMind et complète le modèle AlphaMissense, axé sur la classification des variations des régions codantes des protéines.
Jun Cheng, co-auteur principal de l'article, a déclaré sur son compte X personnel : « Les erreurs d'épissage de l'ARN sont une cause fréquente de nombreuses maladies. Pour la première fois, nous avons construit un modèle unifié capable de prédire simultanément la couverture du séquençage de l'ARN, les sites d'épissage, leur utilisation et les jonctions d'épissage spécifiques qu'ils forment, offrant ainsi une représentation plus complète des résultats d'épissage. » Il a également souligné queL’une des avancées importantes d’AlphaGenome est « la capacité de prédire les jonctions d’épissage directement à partir des séquences et de les utiliser pour prédire les effets des variantes ».
« C'est une étape importante dans le domaine », a déclaré Caleb Lareau, MD, du Memorial Sloan Kettering Cancer Center.Pour la première fois, nous disposons d’un modèle qui combine un contexte long, une précision à base unique et des performances de pointe.DeepMind a ouvert une version préliminaire aux utilisateurs de recherche non commerciaux via l'API AlphaGenome et prévoit de publier officiellement le modèle à l'avenir.
* Lien vers le document de recherche :
Basé sur 1 million de séquences d'ADN et d'informations sur les espèces, en utilisant une conception de type U-Net
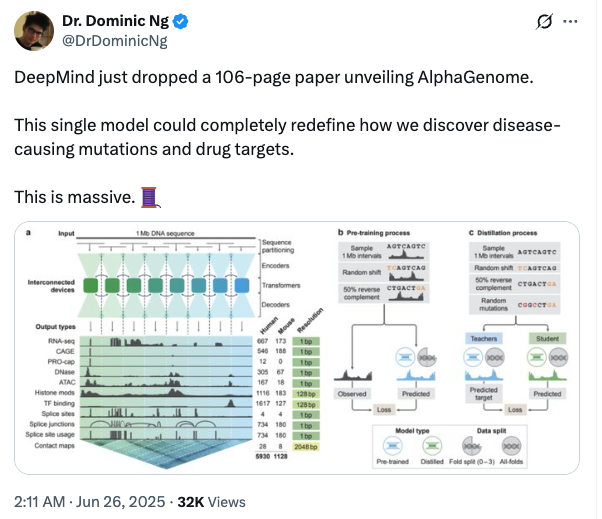
Comme le montre la figure a ci-dessous, le modèle d'apprentissage profond AlphaGenome prend 1 Mb (million de bases) de séquence d'ADN et d'informations sur l'espèce (humaine/souris) en entrée.5 930 loci du génome humain ou 1 128 loci du génome de souris pour la prédiction de différents types de cellulesCouvre 11 types de sortie, notamment :
* Expression génétique (ARN-seq, CAGE, PRO-cap)
* Modèles d'épissage détaillés (sites d'épissage, fréquence d'utilisation des sites d'épissage, jonctions d'épissage)
* État de la chromatine (DNase, ATAC-seq, modification des histones, liaison aux facteurs de transcription)
* Carte de contact de la chromatine
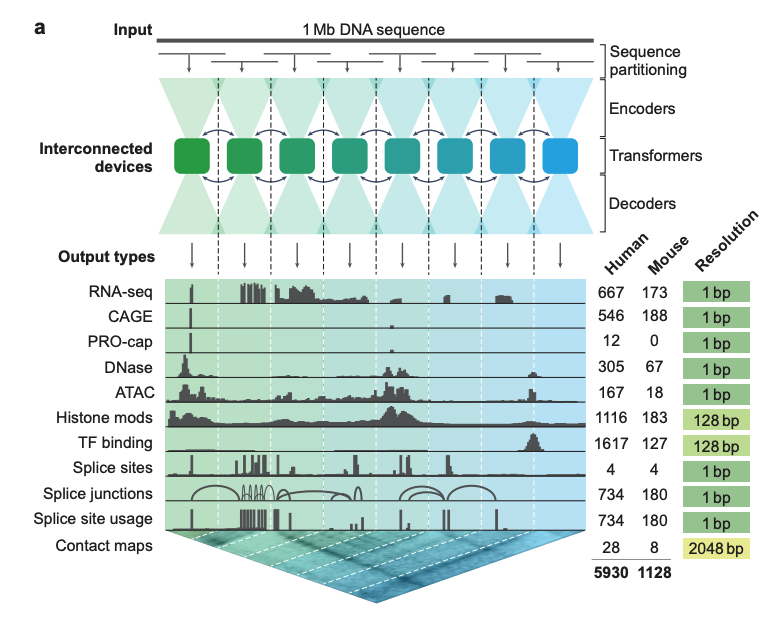
En termes d'architecture de modèle,AlphaGenome adopte une conception d'architecture dorsale de type U-Net.Comme le montre la figure a ci-dessous, la séquence d'entrée peut être efficacement traitée en deux types de représentations de séquence :
Incorporations unidimensionnelles (résolution de 1 pb et 128 pb) : représentent des séquences génomiques linéaires et sont utilisées pour générer des prédictions de trajectoires génomiques.
* Incorporations bidimensionnelles (résolution 2048 pb) : représentent les interactions spatiales entre les fragments génomiques et sont utilisées pour prédire les cartes de contact par paires.
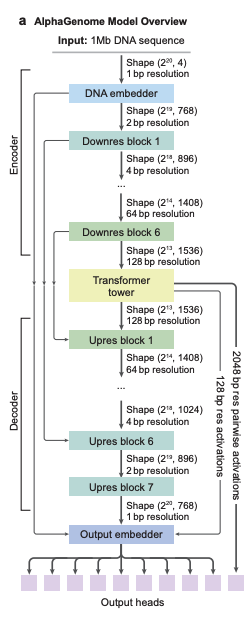
Les couches convolutives du modèle permettent de modéliser des séquences locales afin de réaliser des prédictions fines, tandis que les modules Transformer permettent de modéliser des dépendances à plus long terme, telles que les interactions entre activateurs et promoteurs. Le modèle peut être entraîné sur une base unique, sur une séquence complète de 1 Mo, grâce au parallélisme de séquence distribué, et peut s'exécuter sur huit dispositifs TPUv3 interconnectés.
En termes de formation de modèle,Les chercheurs ont adopté une formation en deux étapes, à savoir la pré-formation et la distillation.Dans la phase de pré-formation, il utilise les données expérimentales existantes pour former deux types de modèles, comme illustré dans la figure b ci-dessous :
* Modèles spécifiques au pliage :L'apprentissage a été réalisé selon une approche de validation croisée en quatre étapes : les trois quarts des segments du génome de référence ont été utilisés pour l'apprentissage et le quart restant pour la validation et les tests. Ces modèles ont permis d'évaluer la capacité d'AlphaGenome à prédire les trajectoires génomiques sur des segments de génome de référence invisibles.
* Modèles tout-plis :Le modèle des enseignants est formé sur tous les segments disponibles du génome de référence comme étape de distillation suivante, comme illustré dans la figure c ci-dessous.
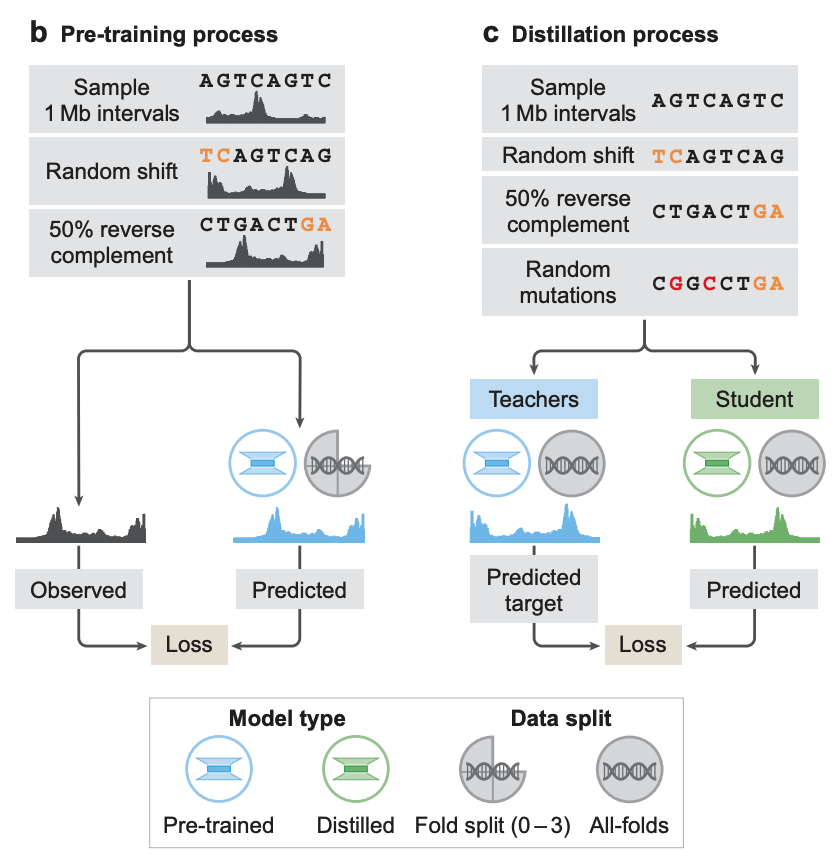
Dans la phase de distillation, les chercheurs ont formé un modèle étudiant qui partageait une architecture pré-entraînée.L’objectif est de prédire la sortie combinée de plusieurs modèles d’enseignants entièrement pliés à l’aide de séquences d’entrée augmentées de manière aléatoire.Des études antérieures ont montré que ce modèle distillé peut simultanément atteindre une plus grande robustesse et une plus grande précision de prédiction des effets variationnels (VEP) dans une instance de modèle.
Grâce à cette conception, le modèle Student peut réaliser la tâche de prédiction des effets de variation de toutes les modalités et types de cellules avec un seul appel d'appareil.Sur un GPU NVIDIA H100, les prédictions pour chaque variante prennent moins d'une seconde.Cela le rend extrêmement efficace pour prédire les effets de variation à grande échelle par rapport aux méthodes d’intégration multi-modèles traditionnelles.
AlphaGenome est leader dans diverses tâches de prédiction du génome
Selon DeepMind, AlphaGenome présente les avantages uniques suivants par rapport aux méthodes existantes :
Contexte de séquence longue + résolution de base unique
AlphaGenome peut analyser des séquences d'ADN jusqu'à un million de bases et effectuer des prédictions à l'échelle d'une base. Cela lui permet de couvrir des régions éloignées de gènes régulateurs tout en capturant des détails biologiques précis. Les modèles précédents se concentraient souvent sur l'équilibre entre la longueur des séquences et la précision des prédictions, limitant ainsi la portée et la précision des modalités modélisables. L'avancée technologique d'AlphaGenome lève cette limite : l'apprentissage utilise la moitié des ressources informatiques du modèle Enformer original et une session d'apprentissage ne prend que 4 heures.
Capacités complètes de prévision multimodale
La combinaison de séquences d'entrée haute résolution et longues permet à AlphaGenome de prédire des modèles de régulation d'une diversité sans précédent, fournissant aux chercheurs des informations plus systématiques sur la régulation des gènes.
Notation efficace des mutations
AlphaGenome peut évaluer l'impact des variants en une seconde. En comparant les différences prédites dans les séquences avant et après le variant et en utilisant la méthode de synthèse la plus adaptée aux différentes modalités, il peut évaluer rapidement et précisément l'impact potentiel des variants génétiques sur les mécanismes moléculaires.
Nouvelle modélisation du site d'épissage
AlphaGenome permet de prédire de manière innovante l'emplacement des jonctions d'épissage de l'ARN et leurs niveaux d'expression directement à partir des séquences. De nombreuses maladies génétiques rares (comme l'amyotrophie spinale et certains types de mucoviscidose) sont associées à des erreurs d'épissage, et cette capacité offre un nouvel outil pour la recherche étiologique.
Excellentes performances dans les benchmarks
AlphaGenome est leader dans diverses tâches de prédiction du génome, telles que la prédiction de la proximité structurale de l'ADN, l'effet des mutations sur l'expression des gènes et les modifications des schémas d'épissage. Il a surpassé les meilleurs modèles existants dans 22 des 24 évaluations de prédiction de séquences d'ADN, et a égalé ou dépassé les meilleurs modèles actuels dans 24 des 26 tâches d'effet de mutation. Plus important encore, c'est aussi le seul modèle capable de réaliser des prédictions conjointes pour toutes les modalités d'évaluation, démontrant ainsi sa grande polyvalence.
Plus précisément, pour évaluer les performances des modèles d’AlphaGenome,Les chercheurs ont d’abord examiné sa capacité de généralisation à des segments génomiques invisibles, ce qui est une condition préalable pour parvenir à une prédiction de haute qualité des effets des variantes.Ils ont réalisé un total de 24 évaluations de prédiction de trajectoire génomique couvrant les 11 modalités prédites par le modèle. Lors des évaluations de validation croisée hors repli, les chercheurs ont utilisé des modèles AlphaGenome pré-entraînés spécifiques au repli et ont comparé leurs prédictions au modèle externe le plus robuste pour chaque tâche.
Les résultats montrent queAlphaGenome a surpassé les modèles externes correspondants dans 22 des 24 évaluations.Comme le montre la figure d ci-dessous. Il convient de noter que dans la tâche de prédiction du changement d'expression génique spécifique au type de cellule (log-fold change, LFC), AlphaGenome a montré une amélioration relative des performances de +17,4% par rapport à un autre modèle de séquence multimodale Barzoï, comme le montre la figure e ci-dessous.
De plus, AlphaGenome a surpassé les modèles spécialisés axés sur une seule modalité pour diverses tâches. Par exemple :
Dans la prédiction de la carte de contact de la chromatine,AlphaGenome surpasse le modèle Orca, comme le montre une augmentation de +6,3% dans le coefficient de corrélation de Pearson de la carte de contact et une augmentation de +42,3% dans les différences spécifiques au type de cellule, comme le montre la figure d ci-dessous ;
Dans la prédiction des pistes du site de démarrage de la transcription,AlphaGenome surpasse ProCapNet, avec une amélioration globale du coefficient de corrélation de Pearson de +15% ;
Dans la prédiction de l'accessibilité de la chromatine,AlphaGenome surpasse ChromBPNet, s'améliorant de +8% sur ATAC-seq et de +19% sur DNase-seq.
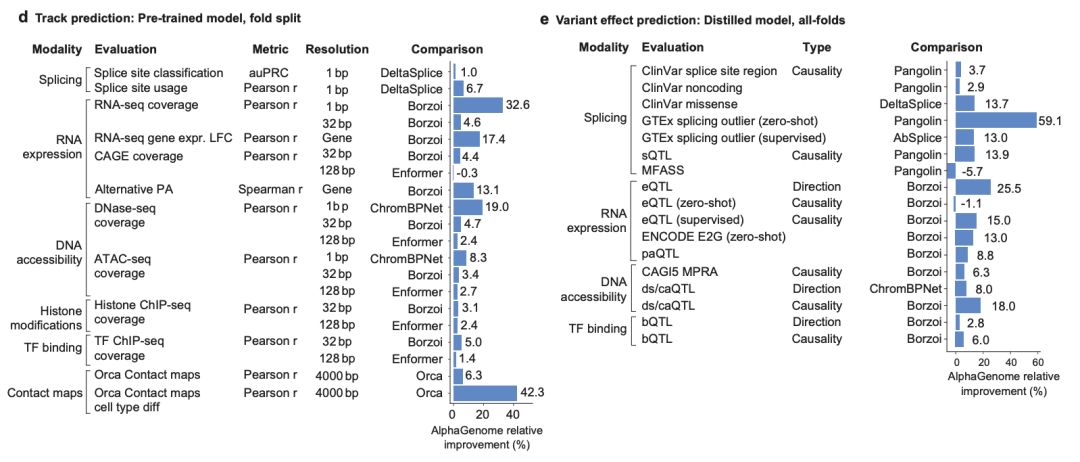
* Figure e : Amélioration relative des performances d’AlphaGenome dans la tâche de prédiction des effets des variantes partielles.
Une étape importante dans l'industrie très appréciée
Le lancement de ce modèle à succès par AlphaGenome continue de susciter de vives discussions sur Twitter depuis la publication de la nouvelle.
Pushmeet Kohli, vice-président de la recherche chez DeepMind, a déclaré : « AlphaGenome fournit une vue complète du génome non codant humain en prédisant l'impact des variantes d'ADN.Cela approfondira notre compréhension de la biologie des maladies et ouvrira de nouvelles voies de recherche. »Dans la section des commentaires, en plus des exclamations et des éloges, tout le monde se soucie davantage de la manière de l'utiliser.
Un doctorant en génétique de l’Université d’Édimbourg a déclaré :« Ce modèle pourrait complètement redéfinir la façon dont nous découvrons les mutations responsables de maladies et les cibles médicamenteuses, ce qui est d’une grande importance. »
Un commentateur dans le domaine des sciences biologiques a déclaré que « AlphaGenome n'est pas seulement un gène unique, mais le génome régulateur entier. Si l'ADN est comparé au code, alors AlphaGenome est le logiciel composé de code. »
En termes d’applications pratiques, AlphaGenome dispose d’un large potentiel de recherche scientifique.Par exemple, face àRecherche sur les mécanismes des maladies,Elle permet de prédire avec plus de précision l'impact des variations génétiques sur les processus de régulation, d'identifier les variations pathogènes potentielles et de révéler de nouvelles cibles. Elle est particulièrement adaptée à l'étude des variations rares ayant des effets significatifs.existerDans le domaine de la biologie synthétique,Diriger la conception de l’ADN pour des fonctions régulatrices spécifiques, telles que l’activation d’un gène cible uniquement dans les cellules nerveuses.existerDans la recherche fondamentale en génomique, il peutAccélérez la localisation et la définition du rôle des éléments fonctionnels clés et aidez à identifier les « instructions de base » nécessaires pour réguler la fonction de types de cellules spécifiques.
Le professeur Marc Mansour de l'University College London a commenté : « AlphaGenome fournit des éléments clés pour l'identification à grande échelle du rôle des variants non codants, nous permettant ainsi de mieux comprendre des maladies complexes comme le cancer. » AlphaGenome est actuellement ouvert à la recherche à des fins non commerciales, et nous attendons avec impatience de nouvelles avancées de la communauté universitaire sur cette base.








