Command Palette
Search for a command to run...
Sélectionné Pour l'ICML 2025, l'Université Tsinghua/Université Renmin a Proposé UniSim, Un Simulateur De Dynamique Biomoléculaire Unifié

Le groupe du professeur Liu Yang de l'Université Tsinghua et le groupe du professeur Huang Wenbing de la Gaoling School of Artificial Intelligence de l'Université Renmin de Chine ont proposé conjointement un simulateur de dynamique biomoléculaire unifié UniSim.Cette méthode obtient un modèle de représentation unifié de tous les atomes grâce à un pré-entraînement hybride de débruitage + champ de force sur une grande quantité de données de structure moléculaire 3D, apprend le champ vectoriel de transfert des molécules à un pas de temps long sur la base d'un cadre génératif interpolant stochastique, et introduit un noyau guidé par la force pour s'adapter rapidement à différents environnements chimiques. UniSim est le premier à réaliser un cadre de simulation dynamique unifié et temporel pour les types moléculaires (petites molécules, peptides, protéines) et les environnements chimiques.Il a favorisé l’application pratique de l’apprentissage profond dans le domaine de la simulation moléculaire.
Les résultats connexes ont été sélectionnés pour l'ICML 2025 sous le titre « UniSim : un simulateur unifié pour la dynamique temporelle des biomolécules ».

Adresse du document :
Autres articles sur les frontières de l'IA :
https://go.hyper.ai/UuE1o
Pourquoi avons-nous besoin d’un simulateur de grossissement temporel unifié ?
Les chercheurs estiment que dans le domaine de la simulation de dynamique moléculaire, il est raisonnable et nécessaire de construire un simulateur de grossissement temporel unifié.D’une part, un cadre de modélisation unifié constitue la base de la simulation collaborative entre les systèmes moléculaires.Par exemple, lors de la simulation de systèmes complexes tels que les interactions protéine-ligand, les protéines et les petites molécules coexistent souvent dans le même environnement physique. Si le modèle ne s'applique qu'à un certain type de molécule, il sera difficile de restituer avec précision le couplage entre les deux à l'échelle atomique. Par conséquent, les simulateurs dotés de capacités de représentation unifiées peuvent gérer simultanément des molécules de types croisés dans le même cadre de modélisation, offrant ainsi une base solide pour la modélisation de complexes multimoléculaires.
D’autre part, un modèle unifié permet d’intégrer les données structurelles et dynamiques de différents types de molécules, améliorant ainsi les capacités de généralisation et de transfert du modèle.Les données de trajectoire moléculaire actuellement disponibles sont très rares et inégalement réparties. Différents types de données, tels que les protéines, les peptides et les petites molécules, présentent des atouts spécifiques. Si elles peuvent toutes participer au pré-apprentissage et à l'apprentissage au sein d'un même modèle, cela améliorera considérablement la compréhension globale des structures atomiques et lui permettra de renforcer ses capacités de migration inter-domaines moléculaires.
en même temps,L’introduction de la simulation de grossissement temporel est également un moyen essentiel d’améliorer l’efficacité de la simulation.Les simulations de dynamique moléculaire traditionnelles s'appuient sur des pas de temps extrêmement courts (de l'ordre de quelques femtosecondes) pour progresser étape par étape, ce qui est coûteux en calcul et rend difficile la prise en compte de comportements à long terme tels que le repliement des protéines. La méthode de grossissement temporel apprend directement la relation de correspondance entre l'état actuel et l'état futur. En préservant la cohérence physique, elle permet de générer rapidement des trajectoires à une échelle de temps bien supérieure aux pas traditionnels, améliorant ainsi considérablement l'efficacité de la simulation et permettant de réaliser des simulations à long terme dans des délais raisonnables.
Représentation unifiée : résoudre le problème de la caractérisation de molécules multi-échelles et multi-types
Bien que les modèles de représentation unifiés de tous les atomes soient la pierre angulaire des simulations dynamiques à travers les espèces moléculaires,Cependant, la mise en œuvre d’un tel modèle se heurte encore aux défis techniques suivants :
* Premièrement, les systèmes moléculaires vont des petites molécules organiques de plusieurs dizaines d’atomes aux macromolécules protéiques de milliers d’atomes, avec d’énormes différences d’échelle et des structures complexes et diverses.Si tous les atomes sont utilisés directement pour l’entraînement, le modèle aura des mécanismes d’attention différents pour différents types de molécules, inhibant ainsi la capacité de transfert croisé du modèle.
* Deuxièmement, la base pour parvenir à une représentation unifiée de tous les atomes est d’utiliser un vocabulaire unifié au niveau atomique.Une approche intuitive consiste à utiliser directement le tableau périodique comme vocabulaire pour l'intégration de la représentation. Cependant, cette approche ignore les unités régulières présentes en grand nombre dans les peptides et les protéines, telles que les sous-structures comme les acides aminés naturels, ce qui entraîne de faibles performances sur les données de type protéique.
* Enfin, afin d'apprendre pleinement la représentation des molécules dans différents états, une grande quantité de données de structure moléculaire 3D à l'état stationnaire et non stationnaire sera incluse dans l'ensemble de données de pré-formation.Le paradigme courant pour le pré-entraînement des molécules instables consiste à apprendre les forces agissant sur les atomes. Cependant, les différents jeux de données utilisent des paramètres de champ de force différents lors du calcul des champs de force atomiques, ce qui entraîne un décalage des données d'étiquetage.
Afin de parvenir à une modélisation unifiée, UniSim introduit trois technologies clés pour résoudre les problèmes ci-dessus :
* Sous-graphe gradient-environnement : équilibrer raisonnablement l'échelle moléculaire
Dans l’étape de prétraitement des données, les données de structure 3D des grandes molécules (avec plus de 1 000 atomes) seront segmentées.min < rmax Lors du prétraitement, n'importe quel atome de la molécule sera sélectionné au hasard et l'atome sera considéré comme le centre de la sphère.min et rmax Faire une sphère avec un rayonLes atomes contenus dans la petite boule sont considérés comme le sous-graphe du gradient, et les atomes contenus dans la grande boule sont considérés comme le sous-graphe de l'environnement.Sur la base de la théorie physique selon laquelle la force interatomique décroît généralement de manière exponentielle avec la distance, lorsque rmax– rmin Lorsqu'il est correctement sélectionné, l'interaction entre les atomes extérieurs au sous-graphe d'environnement de la molécule d'origine et ceux du sous-graphe de gradient sera négligeable. Par conséquent, lors de l'apprentissage, le sous-graphe d'environnement sera utilisé comme entrée à la place de la molécule d'origine, et seul le sous-graphe de gradient sera impliqué dans le calcul de la fonction de perte, ce qui équilibre raisonnablement l'échelle des données de structure moléculaire et améliore la capacité de transfert croisé du modèle.
* Extension d'intégration d'atomes : obtenez une représentation atomique plus raffinée
Cette étude est basée sur le tableau périodique des éléments.Présentation de plusieurs représentations d'intégration discrètes apprenables pour le même élément sous forme de vocabulaire étendu,Il permet de capturer la sous-structure régulière où se situent les atomes. Basé sur un réseau neuronal graphique simple, UniSim intègre les informations de voisinage de chaque atome, calcule la probabilité de chaque représentation intégrée dans le vocabulaire étendu correspondant à l'atome et obtient la représentation intégrée étendue de l'atome par sommation pondérée.Cette représentation équilibre la précision au niveau atomique avec des sous-structures régulières au sein d'espèces moléculaires spécifiques, ce qui donne une représentation atomique efficace et détaillée.
* Pré-entraînement hybride multi-têtes : apprentissage hybride de données avec différents états moléculaires et distributions d'étiquettes
UniSim utilise la méthode suivante pour apprendre conjointement les structures moléculaires à l'état stationnaire et non stationnaire : pour les données à l'état stationnaire, l'article utilise le paradigme de pré-formation de débruitage pour débruiter les données bruyantes afin d'apprendre la représentation atomique ; pour les données à l'état non stationnaire, le modèle apprendra directement le champ de force conservateur, et différents paramètres de champ de force correspondent à différentes têtes de sortie, évitant ainsi les erreurs introduites par différentes distributions d'étiquettes.
Cet article utilise TorchMD-NET comme modèle de réseau neuronal graphique de base satisfaisant l'équivariance SO(3). En s'appuyant sur les techniques de pré-apprentissage clés décrites ci-dessus,Une pré-formation sur les données moléculaires 3D multi-sources à grande échelle a été réalisée et la construction efficace d'un modèle de représentation atomique unifié a été réalisée.
Modèles de champs vectoriels : apprentissage des transitions d'état à long terme à partir de trajectoires
Les simulations de dynamique moléculaire traditionnelles sont limitées par des étapes d'intégration de quelques femtosecondes, ce qui rend difficile l'échantillonnage efficace de comportements à long terme tels que le repliement des protéines. UniSim adopte un cadre d'interpolation stochastique et connecte un perceptron vectoriel géométrique en tant que modèle de champ vectoriel après un modèle de représentation tout-atome pré-entraîné.Le modèle réalise une modélisation dynamique temporellement grossière de bout en bout en apprenant le champ vectoriel de transfert entre les états moléculaires à de longs pas de temps.
Lors de l'apprentissage, des paires de conformations moléculaires séparées par un pas de temps donné dans la trajectoire dynamique réelle sont sélectionnées comme échantillons d'apprentissage, des perturbations aléatoires sont introduites sur le chemin d'interpolation, et le champ de vitesse (vitesse) et le débruiteur (débruiteur) sont appris conjointement pour générer une trajectoire en temps continu. Comparé à l'intégration numérique traditionnelle, UniSim peut améliorer considérablement l'efficacité de la simulation et surmonter le goulot d'étranglement de la simulation traditionnelle en termes d'échelle de temps.
Noyaux guidés par force : adaptation rapide aux environnements chimiques complexes
La dynamique moléculaire dans différentes conditions de solvant, de température et de pression présente différentes surfaces d'énergie potentielle, qui affectent grandement la distribution des conformations générées.À cette fin, UniSim introduit un noyau de guidage de force pour définir un champ de force intermédiaire virtuel sur le cadre de différence aléatoire pour guider l'échantillonnage de trajectoire.Ce champ de force intermédiaire est équivalent au champ de force MD réel aux deux extrémités du chemin de génération (c'est-à-dire l'état initial et l'état final), et est conçu pour être hautement cohérent avec les priors physiques, de sorte que la conformation générée est plus cohérente avec la distribution de Boltzmann sous le champ de force cible.
En ajustant le champ de force intermédiaire, UniSim n'a pas besoin de modifier les paramètres du modèle pré-entraîné et du modèle de champ vectoriel.Il suffit d'apprendre un noyau de guidage de force enfichable pour que le champ de force cible s'adapte efficacement aux nouveaux environnements chimiques.Améliore efficacement les capacités de généralisation et de migration du modèle.
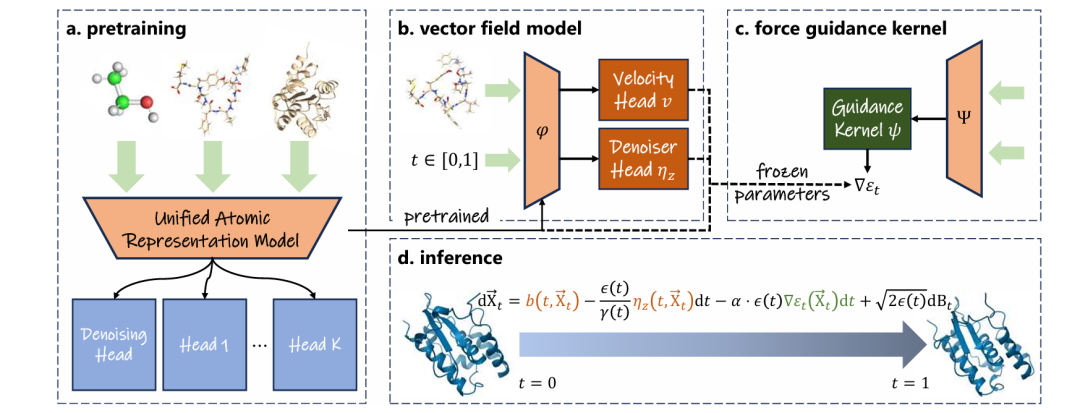
Vérification expérimentale : plusieurs types de molécules
Afin de vérifier la polyvalence d'UniSim sur différents types moléculaires,Les chercheurs ont évalué systématiquement les données de plusieurs types moléculaires dans une tâche de simulation directe, comprenant trois types de molécules : les petites molécules, les peptides et les protéines.En comparant avec le modèle d'apprentissage profond sur le terrain qui effectue également une simulation de dynamique grossière dans le temps, l'expérience vise à explorer si la représentation atomique unifiée peut aider à améliorer la compréhension du modèle des états moléculaires et des capacités de généralisation intermodale, et comment l'implication des noyaux guidés par la force affecte les performances du modèle dans des indicateurs clés tels que la rationalité des conformations générées sous le champ de force cible et la similarité de la distribution.
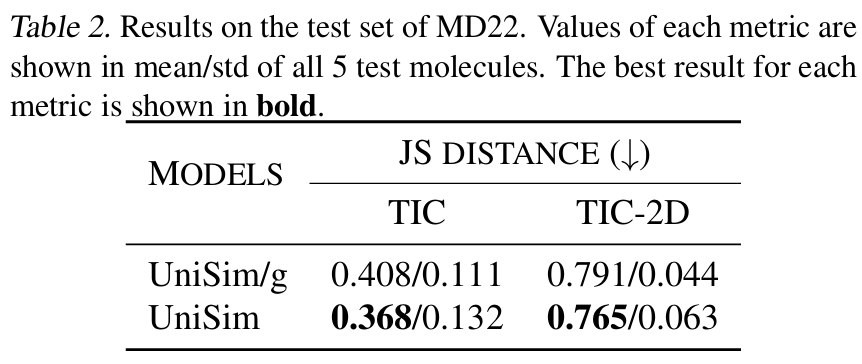
Les résultats montrent qu’UniSim a atteint une supériorité globale dans tous les types moléculaires.Il présente de bons résultats en termes de similarité de distribution et une amélioration significative de l'indicateur clé de rationalité conformationnelle (Val-CA). Il convient de noter que dans la tâche de génération de simulation directe, chaque conformation de la trajectoire est générée par autorégression, ce qui entraîne une erreur cumulative importante ; il est donc assez difficile d'améliorer la rationalité de la conformation.
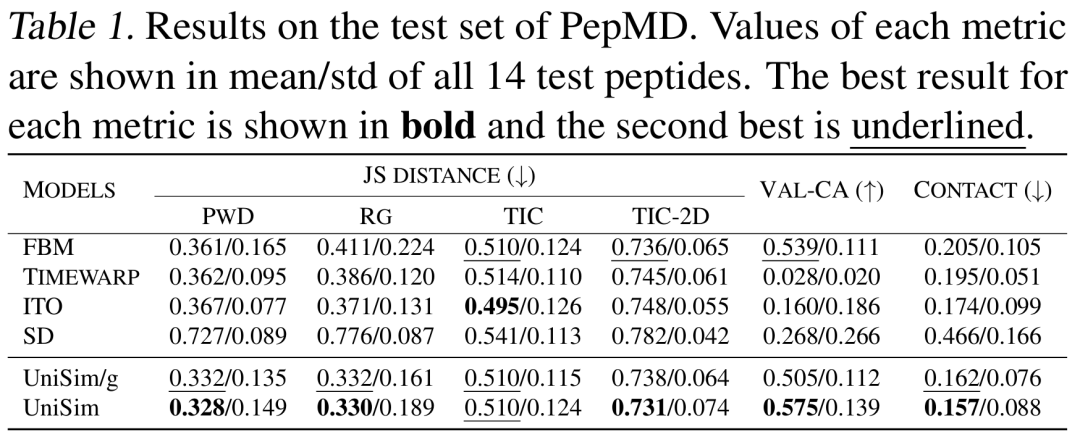
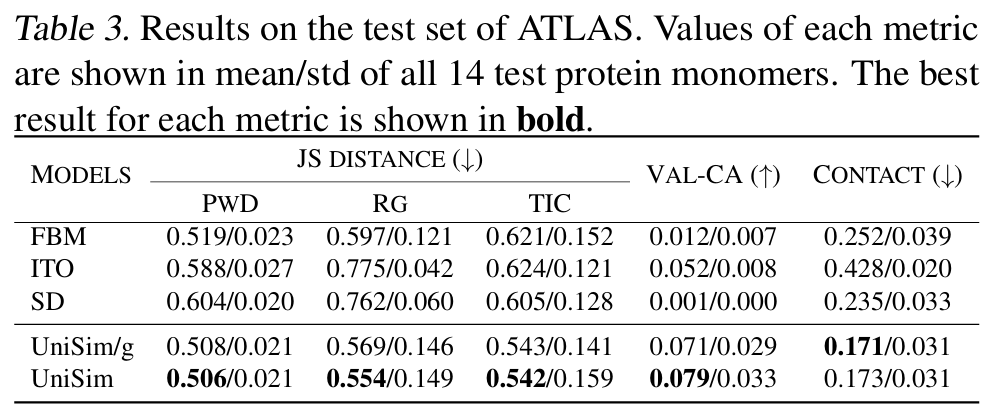
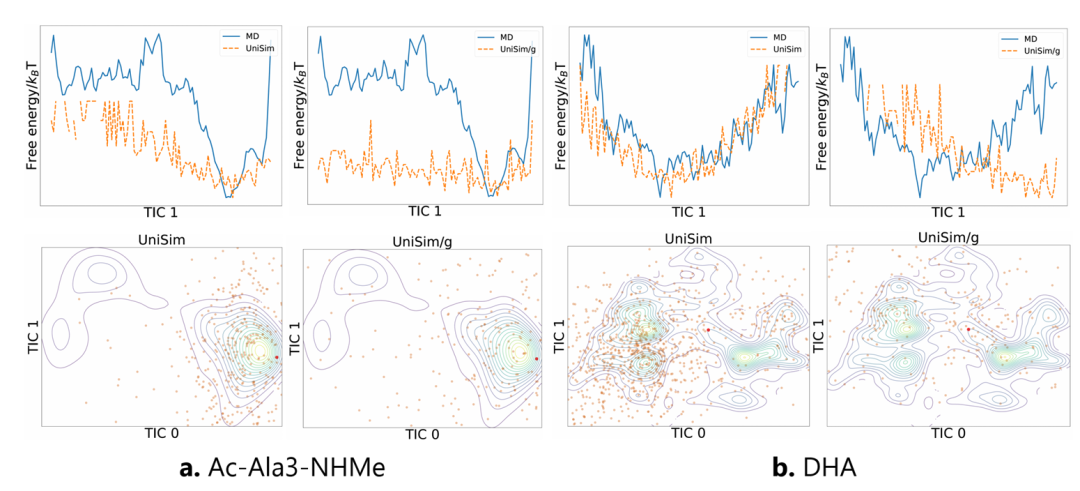
Dans la tâche de simulation directe des peptides et des protéines,Comparé aux méthodes existantes telles que FBM, ITO et SD, UniSim est en avance sur des indicateurs tels que la similarité de distribution (TIC-2D), la rationalité structurale (VAL-CA) et l'erreur de contact (CONTACT). En particulier, après l'introduction du noyau guidé par force, UniSim maintient son niveau initial pour des indicateurs tels que la similarité de distribution, mais a considérablement amélioré les indicateurs clés de rationalité conformationnelle. Parallèlement, dans les systèmes protéiques complexes, UniSim peut franchir les barrières énergétiques et couvrir plusieurs états métastables en seulement quelques centaines d'étapes de simulation directe, ouvrant ainsi de nouvelles perspectives pour une simulation efficace des grandes biomolécules.
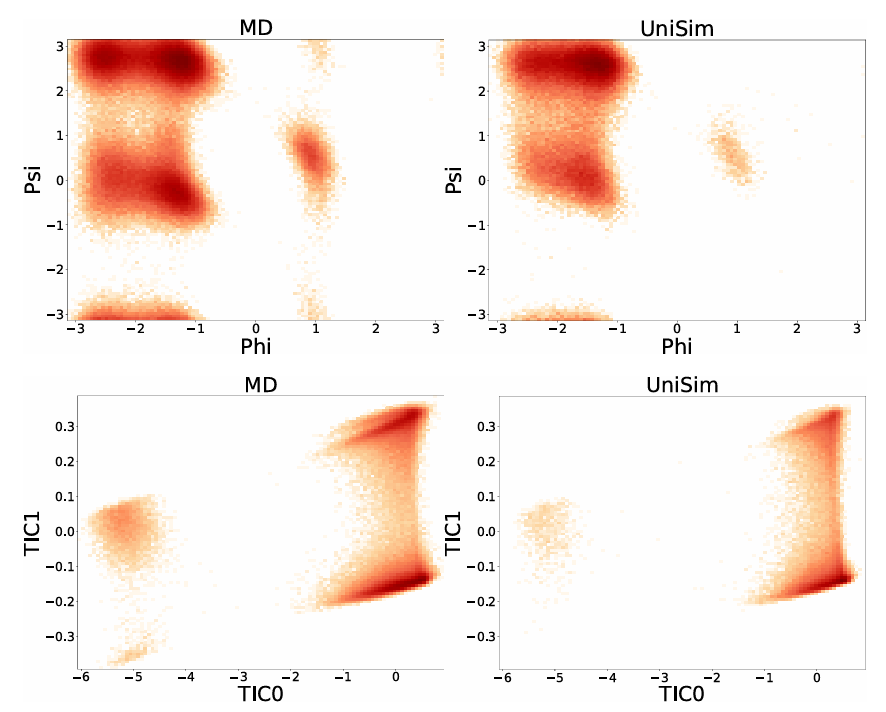
Étude de cas sur l'alanine-dipeptide
De plus, pour explorer la stabilité d'UniSim dans les simulations de dynamique moléculaire à long terme, les chercheurs ont affiné le modèle sur le système classique alanine-dipeptide et effectué des simulations à long terme de 100 000 étapes.En comparant avec les résultats MD, UniSim a reproduit avec succès 5 états métastables clés connus.Le paysage énergétique libre du dipeptide d'alanine dans le processus dynamique a été restauré avec précision, vérifiant pleinement la stabilité et la cohérence physique du modèle dans le cadre d'une simulation à long terme.
Perspectives
UniSim est le premier framework permettant de réaliser une simulation dynamique unifiée à gros grains temporels sur tous les types moléculaires et environnements chimiques.Cela ouvre la voie à une application généralisée de l'apprentissage profond dans la découverte de médicaments, la conception de protéines et d'autres scénarios. Les chercheurs ont également souligné que les pistes suivantes pourraient être explorées plus avant à l'avenir :
* Un mécanisme d’optimisation de conformation intermodale plus efficace pour améliorer l’efficacité des échantillons générés ;
* Modélisation de trajectoires sur des échelles de temps plus longues pour révéler des mécanismes biophysiques complexes ;
* Explorer les mécanismes dynamiques dans les systèmes complexes, en se concentrant sur les interactions intermoléculaires.








