Command Palette
Search for a command to run...
L'Université Du Shandong Développe Un Algorithme d'apprentissage Profond Interprétable RetroExplainer Pour Identifier La Voie De Rétrosynthèse Des Composés Organiques En 4 Étapes

La rétrosynthèse vise à trouver une série de réactifs appropriés pour synthétiser efficacement le produit cible. Il s’agit d’une méthode importante pour résoudre les voies de synthèse organique, et c’est également la méthode la plus simple et la plus basique pour concevoir des voies de synthèse organique.
Les premières recherches rétrosynthétiques reposaient principalement sur la programmation, mais ce travail a ensuite été repris par l’IA. Cependant, les méthodes de rétrosynthèse existantes se concentrent principalement sur la rétrosynthèse en une seule étape, ont une faible interprétabilité et ne peuvent pas prendre en compte les informations à courte et à longue portée des molécules, ce qui entraîne des performances limitées.
À cette fin, Wei Leyi de l'Université du Shandong et le groupe de recherche de Zou Quan de l'Université des sciences et technologies électroniques de Chine ont développé conjointement RetroExplainer. Cet algorithme d'apprentissage profond explicable peut identifier les voies de rétrosynthèse des composés organiques en 4 étapes et fournir des réactifs facilement disponibles. RetroExplainer devrait fournir un outil puissant pour la recherche rétrosynthétique en chimie organique.
Auteur | Xuecai
Rédacteur | Sanyang
La rétrosynthèse en chimie organique vise à trouver une série de réactifs appropriés pour synthétiser efficacement le produit cible.. Ce procédé est un travail de base indispensable en synthèse assistée par ordinateur.
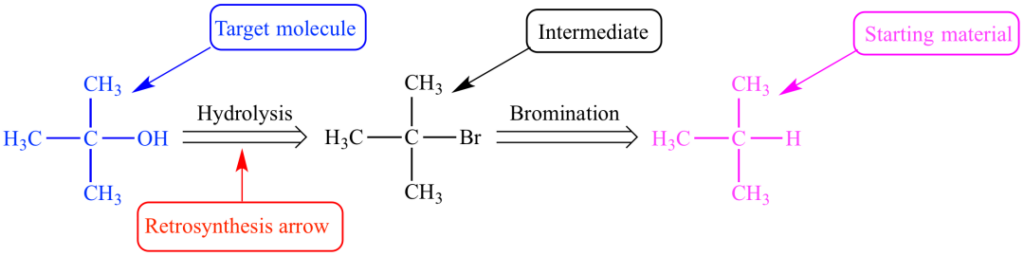
Figure 1 : Voie de rétrosynthèse de l'alcool tert-butylique
Dans les années 1960,Corey et al. a tenté d'effectuer une analyse rétrosynthétique via la programmation, et a développé le logiciel Organic Chemical Simulation Synthesis (OCSS). Cependant,À mesure que la quantité de données augmente, ce travail est rapidement repris par l’IA. Parmi eux, le modèle d’apprentissage profond est très attendu et a produit des résultats considérables.
Dans les premières recherches sur la rétrosynthèse de l'IA, les chercheurs travaillaient souvent à rebours, des produits aux réactifs, en fonction de modèles de réaction, c'est-à-dire une rétrosynthèse basée sur des modèles.. Parmi eux, les empreintes moléculaires basées sur des perceptrons multicouches sont souvent utilisées pour le codage de produits et la sélection de modèles.
Alors,Les chercheurs ont commencé à explorer les méthodes de synthèse sans modèle et semi-modèle, comprenant principalement :
1. Rétrosynthèse basée sur des séquences ;
2. Rétrosynthèse basée sur des diagrammes.
La principale différence entre les deux réside dans la forme sous laquelle les molécules sont exprimées. Le premier utilise des chaînes linéarisées pour représenter les molécules, comme la spécification SMILES ; tandis que ce dernier utilise des modèles de graphes moléculaires pour représenter les molécules, incluant principalement la prédiction des centres de réaction (RC) et l'achèvement des synthons.
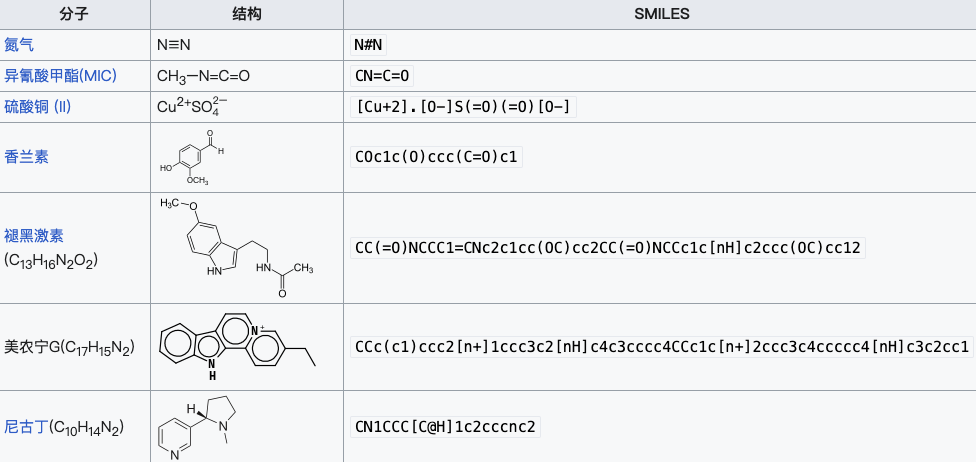
Figure 2 : Expressions SMILES de certaines substances
Bien que les méthodes rétrosynthétiques existantes aient fait des progrès significatifs,Il reste cependant trois problèmes endogènes:
1.La rétrosynthèse basée sur la séquence manque d'informations moléculaires, tandis que la rétrosynthèse basée sur le graphique ignore les informations de séquence et les caractéristiques à longue portée des molécules. Les deux méthodes sont limitées en termes d’apprentissage des fonctionnalités et il est difficile d’améliorer les performances ;
2.Les méthodes de rétrosynthèse basées sur l'apprentissage profond ont une faible interprétabilité. Bien que la rétrosynthèse basée sur un modèle puisse fournir une voie de synthèse compréhensible, le mécanisme de prise de décision de l'algorithme est encore vague, et la reproductibilité et la faisabilité du modèle doivent être prises en compte ;
3.Les méthodes existantes se concentrent principalement sur la rétrosynthèse en une seule étape. Cette approche semble donner des réactifs raisonnables, mais ces réactifs peuvent être difficiles à acheter ou nécessiter un traitement compliqué. Par conséquent, la rétrosynthèse en plusieurs étapes peut être plus significative dans la synthèse chimique réelle.
à cette fin,Wei Leyi de l'Université du Shandong et le groupe de recherche de Zou Quan de l'Université des sciences et technologies électroniques de Chine ont développé conjointement RetroExplainer . Cet algorithme peut effectuer des prédictions rétrosynthétiques basées sur l'apprentissage profond tout en prenant en compte l'interprétabilité et la faisabilité de l'algorithme. RetroExplainer surpasse les autres algorithmes dans près de 12 ensembles de données de référence, et 86,91 % des réactions dans les voies de synthèse proposées de TP3T sont vérifiées par la littérature. Ce résultat a été publié dans Nature Communications.

Ce résultat a été publié dans Nature Communications.
Lien vers l'article :
https://www.nature.com/articles/s41467-023-41698-5
Suivez le compte officiel et répondez « retrosynthesis » pour obtenir le PDF complet de l'article.
Procédures expérimentales
Construction d'algorithmes:Module + Sous-réseau
L'ensemble du processus d'analyse rétrosynthétique comprend quatre étapes : le codage graphique moléculaire, l'apprentissage multitâche, la prise de décision et la prédiction de la voie de synthèse en plusieurs étapes.
RetroExplainer se compose principalement de quatre modules : Multi-sensory Multi-scale Graph Transformer (MSMS-GT), Dynamic Adaptive Multi-task Learning (DAMT), Explainable Decision Module et Route Prediction Module.
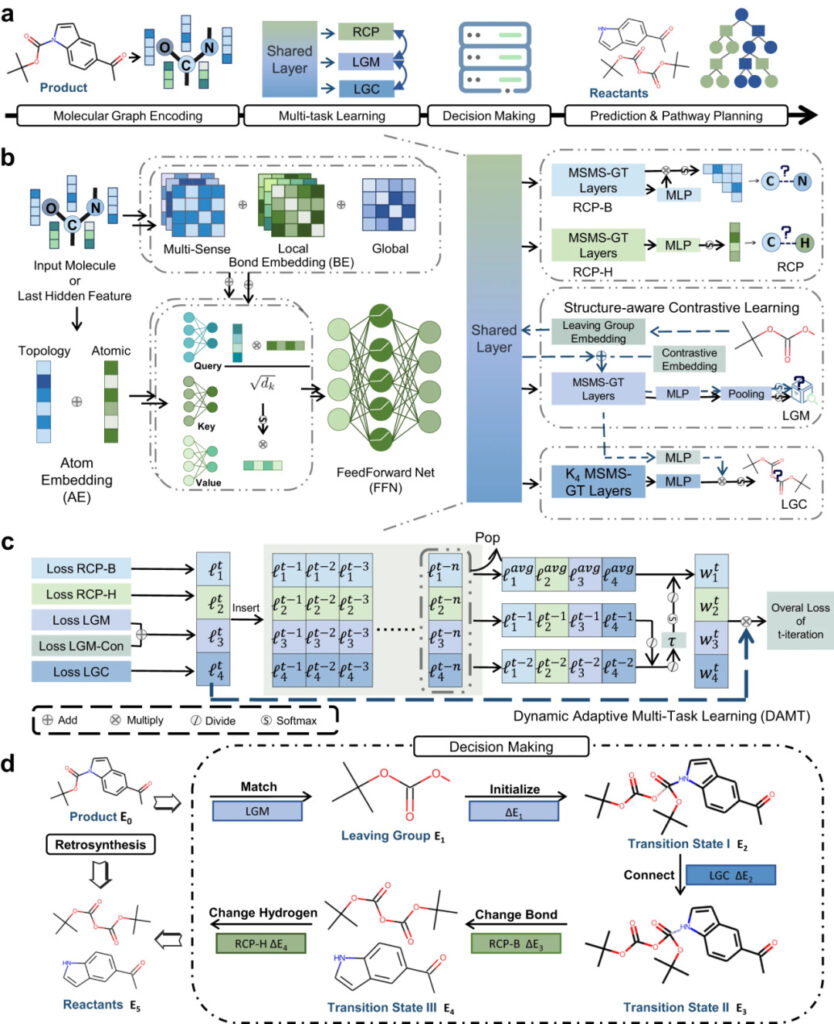
Figure 3 : Schéma de RetroExplainer et de ses modules
a : Diagramme de processus RetroExplainer ;
b : Architecture MSMS-GT ;
c : Schéma de principe de l'algorithme DAMT ;
d : Processus de prise de décision similaire au mécanisme de réaction.
MSMS-GT capture des informations chimiques importantes grâce à l'intégration de liaisons chimiques et à l'intégration topologique d'atomes. Les informations codées sont fusionnées dans des vecteurs moléculaires grâce au mécanisme d'attention multi-têtes.
Dans le module DAMT, les informations moléculaires sont simultanément entrées dans les sous-grilles de prédiction du centre de réaction (RCP), de correspondance du groupe partant (LGM) et de connexion du groupe partant (LGC).
Le RCP identifie les changements dans les liaisons chimiques et le nombre d'atomes d'hydrogène adjacents aux atomes, le LGM fait correspondre le groupe partant du produit à celui de la base de données et le LGC relie le groupe partant au résidu du produit.
Le module de décision convertit les produits en réactifs en fonction des cinq actions rétrosynthétiques et du score énergétique (E) de la courbe de décision, et simule inversement le processus d'assemblage moléculaire.
Enfin, un algorithme de recherche arborescente heuristique est utilisé pour trouver une voie de synthèse de produit efficace tout en garantissant la disponibilité des réactifs.
Comparaison des performances:Ensemble de données de référence de l'USPTO
Pour vérifier les performances de RetroExplainer, les chercheurs l'ont comparé à 21 autres algorithmes de rétrosynthèse basés sur des réactions chimiques incluses dans l'Office des brevets et des marques des États-Unis (USPTO), l'indicateur d'évaluation étant la précision top-k.
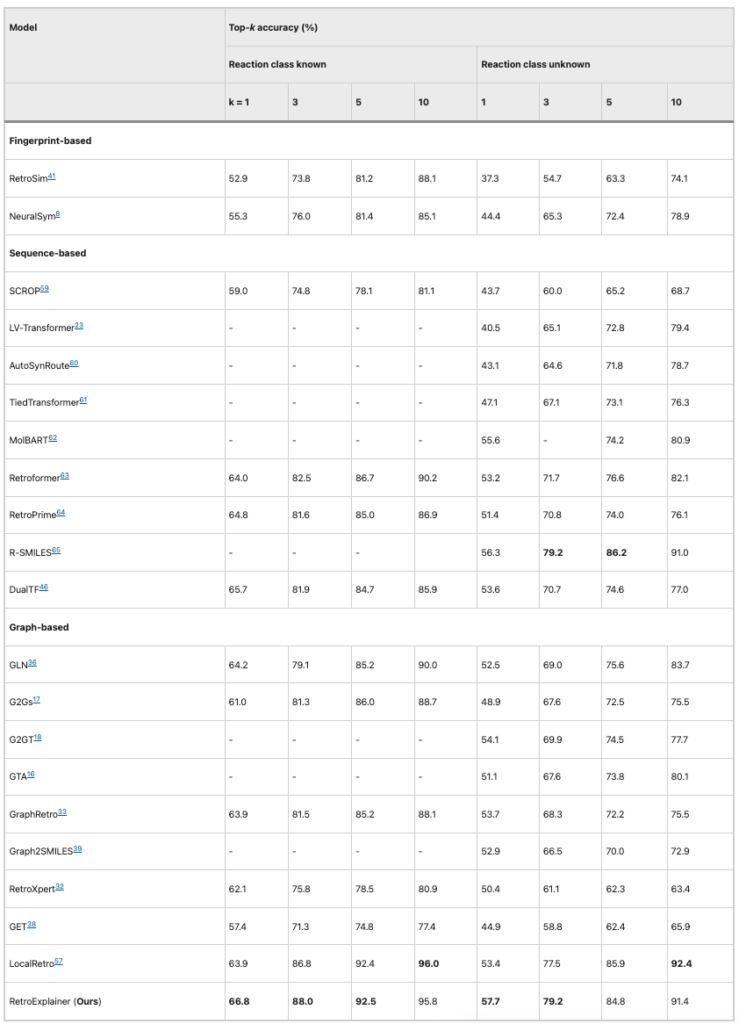
Tableau 1 : Comparaison des performances de RetroExplainer et d’autres algorithmes (USPTO-50K)
On peut constater que sur la base de l’ensemble de données USPTO-50K, parmi les 8 indicateurs d’évaluation,RetroExplainer surpasse les autres algorithmes dans 5 aspects et se classe premier en termes de précision moyenne. Bien que RetroExplainer soit inférieur à LocalRetro en termes de précision top-10, l'écart entre les deux n'est que de 1%.
Pour éliminer l’impact des molécules similaires, les chercheurs ont réparti les données à l’aide de Tanimoto Similarity et les ont comparées aux deux algorithmes les plus précis, R-SMILE et LocalRetro.
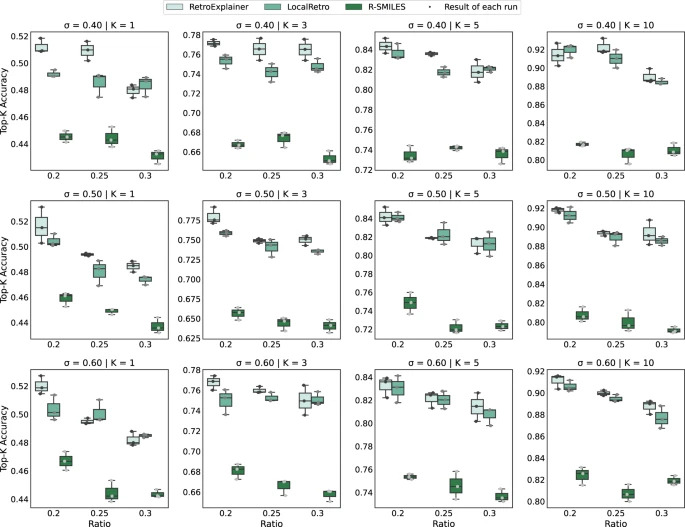
Figure 4 : Comparaison des performances de RetroExplainer, R-SMILES et LocalRetro sur différents ensembles de données
Comme le montrent les résultats, RetroExplainer fonctionne mieux dans la plupart des ensembles de données, ce qui reflète sa stabilité et son adaptabilité.
Les chercheurs ont ensuite comparé les performances de l’algorithme sur les ensembles de données plus vastes de l’USPTO-MIT et de l’USPTO-FULL. RetroExplainer surpasse les autres algorithmes dans tous les indicateurs, et l'écart avec les autres algorithmes est encore plus grand.Cela montre que RetroExplainer a un plus grand potentiel dans l’analyse de données à grande échelle.
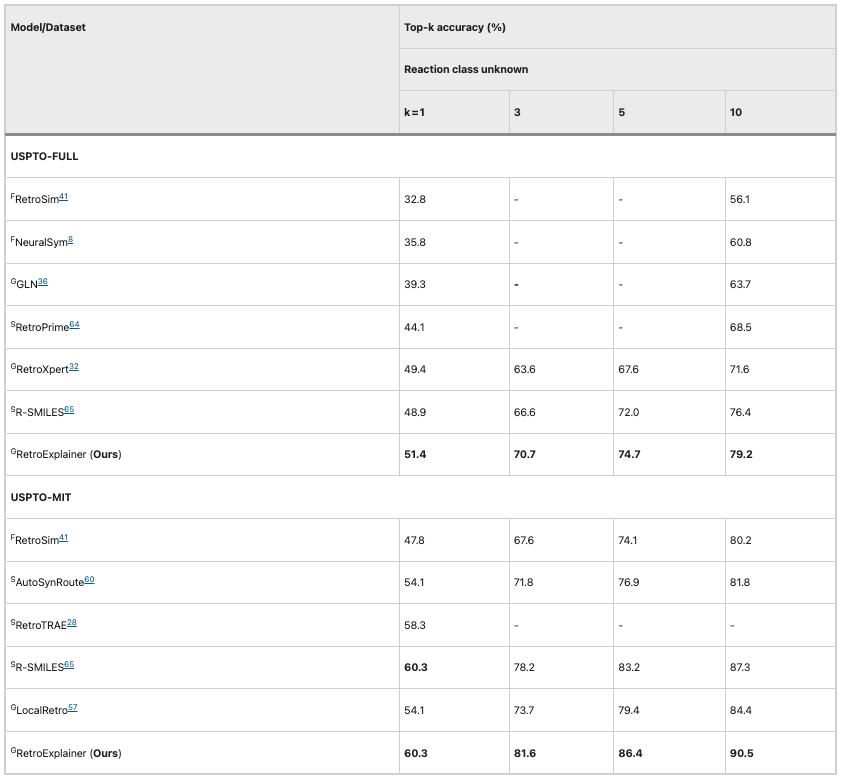
Tableau 2 : Comparaison des performances de RetroExplainer et d’autres algorithmes (USPTO-MIT et USPTO-FULL)
Explicabilité:Visualisation des décisions
Inspirés par la réaction de substitution nucléophile bimoléculaire (SN2), les chercheurs ont conçu un processus de prédiction rétrosynthétique interprétable basé sur l'assemblage moléculaire guidé par l'apprentissage profond. Le processus de prise de décision comprend six étapes : le produit d'origine (P), la correspondance du groupe partant (S-LGM), l'initialisation (IT), la connexion du groupe partant (S-LGC), le changement de liaison chimique du centre de réaction (S-RCP) et le changement du nombre d'atomes d'hydrogène (HC).
En fonction de la contribution de chaque étape à la décision finale, le sous-réseau DAMT génère un score énergétique (E) pour chaque étape.
Le processus spécifique est le suivant :
1. Dans l'étape P, E de chaque étape est initialisé à 0 ;
2. Dans l’étape S-LGM, le groupe partant est sélectionné en fonction de la probabilité prédite du module LGM ;
3. Ajoutez l'E du groupe partant sélectionné dans l'étape S-LGM à la probabilité de l'événement de réponse prédite par les modules RCP et LGM pour obtenir l'énergie de l'étape IT ;
4. Dans les étapes S-LGC et S-RCP, tous les nœuds possibles dans l'arbre de recherche sont développés en fonction de l'algorithme de programmation dynamique. Sélectionnez les événements avec une probabilité supérieure à un seuil prédéfini et gardez E fixe ;
5. Ajustez le nombre d'atomes d'hydrogène et la charge formelle de chaque atome pour vous assurer que le diagramme moléculaire résultant est conforme aux règles de valence et calculez le E final.
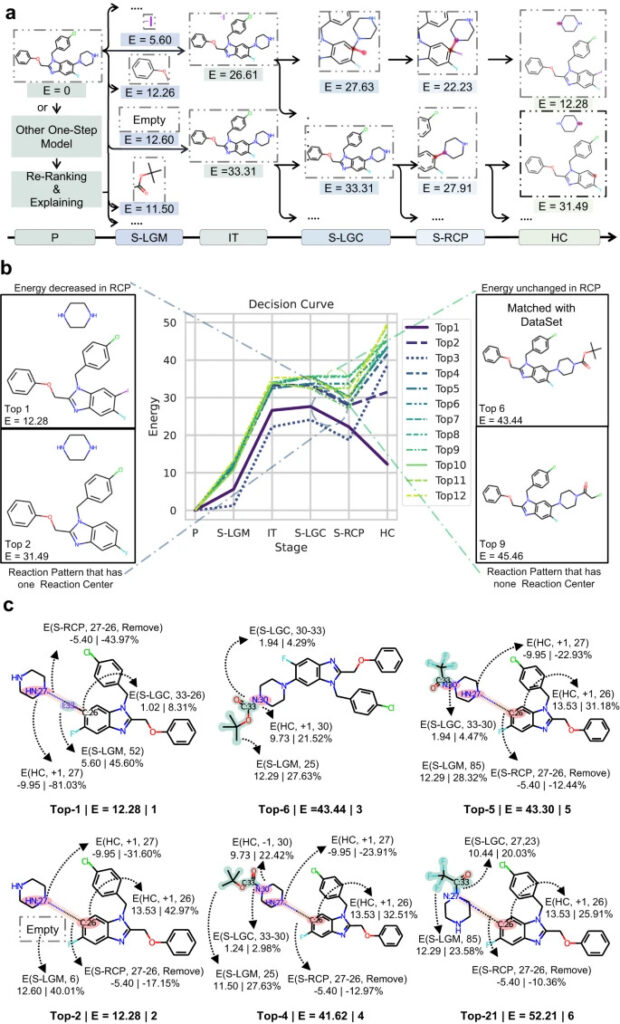
Figure 5 : Processus décisionnel de RetroExplainer
a : chemin de recherche de RetroExplainer pour deux résultats de prédiction ;
b : Courbes de décision des 12 principaux itinéraires prédits ;
c : 6 processus de changement structurel représentant les voies de synthèse.
En traçant une courbe de décision basée sur le changement de E, nous pouvons analyser le processus de décision de RetroExplainer et découvrir les erreurs de prédiction de RetroExplainer.
Comme le montre la figure, la voie de synthèse correcte vers le produit devrait être la réaction de déprotection de l'amine, mais RetroExplainer la classe 6ème, et la 1ère est la réaction de couplage CN. L’analyse a révélé que RetroExplainer avait tendance à augmenter le nombre d’atomes d’hydrogène des amines dans l’étape HC, ce qui a conduit à cette différence.Cela indique que RetroExplainer peut faire la même erreur de jugement sur des molécules ayant des structures similaires au stade HC.
En comparant les réactions de RetroExplainer classées 1er et 2ème,Les chercheurs ont découvert que E pourrait être lié à la difficulté de la réponse. Bien que la connexion de I:33 et C:26 dans la réaction 1 ne soit pas propice à la réduction d'énergie, la connexion d'un atome d'hydrogène à C:26 nécessite 13 fois l'énergie de la réaction précédente. Dans le même temps, l’introduction de I:33 a affaibli le problème de sélectivité auquel est confrontée la réaction de couplage CN.
en même temps,L'encombrement stérique peut également affecter les résultats de prédiction de RetroExplainer. En comparant les réactions classées 4e et 21e, leurs structures moléculaires sont les mêmes, mais les groupes partants sont attachés au N symétrique, ce qui entraîne des différences dans E.
Planification du parcours:Voies de synthèse prédites en plusieurs étapes
Pour améliorer la praticité des prédictions de RetroExplainer, les chercheurs l'ont combiné avec l'algorithme Retro, remplaçant les prédictions en une seule étape de ce dernier par des prédictions en plusieurs étapes.
En prenant comme exemple le bronchodilatateur Protokylol, RetroExplainer a conçu une voie de synthèse en 4 étapes pour ce produit. Les chercheurs ont ensuite mené une revue de la littérature sur ces réactions en quatre étapes pour explorer leur faisabilité.
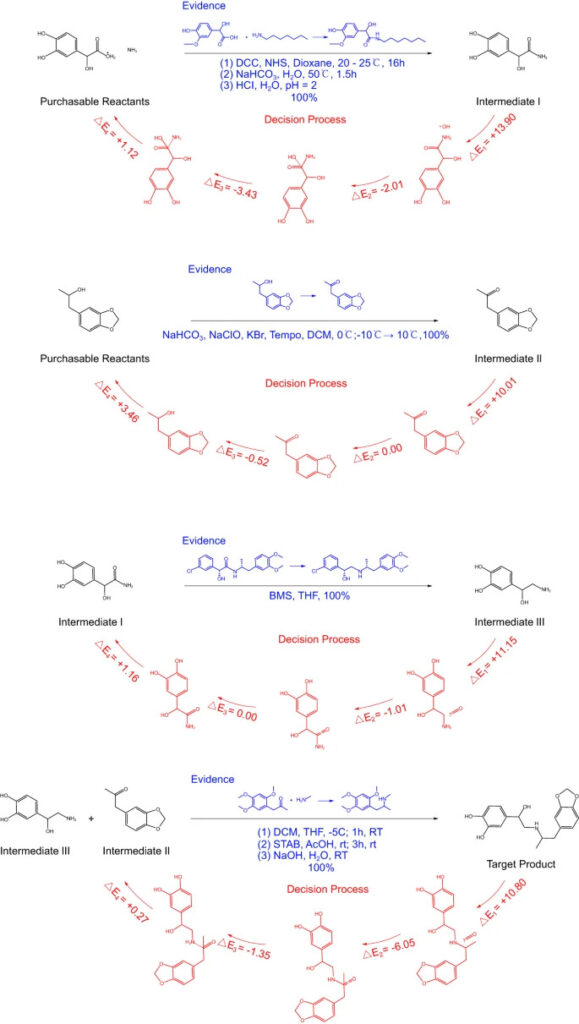
Figure 6 : Synthèse du protosane en 4 étapes par RetroExplainer
Le texte bleu dans la figure est une réponse similaire enregistrée dans la référence, et la partie rouge est le processus de prise de décision de RetroExplainer.
Bien que de nombreuses réactions n’aient pas trouvé exactement la même référence, elles ont trouvé des réactions similaires à haut rendement. aussi,RetroExplainer a conçu 176 expériences pour 101 cas, dont 153 ont pu trouver des réactions similaires dans SciFinder.
Les résultats ci-dessus montrent que la prédiction de la rétrosynthèse de RetroExplainer est meilleure que celle des autres algorithmes actuels. Dans le même temps, les décisions de RetroExplainer sont transparentes et explicables, et il effectue une planification en plusieurs étapes pour les réponses, ce qui les rend plus réalisables. RetroExplainer devrait fournir un outil puissant pour la recherche rétrosynthétique en chimie organique.
Performance vs explicabilité, la contradiction de l'IA
L'explicabilité est un facteur clé dans l'application de l'IA dans divers scénarios. Avec le développement continu de l’IA dans des secteurs tels que la conduite autonome, le diagnostic médical, la finance et les assurances, le processus de prise de décision de l’IA est devenu de plus en plus important et est confronté à de plus en plus de problèmes pratiques, sociaux et même juridiques.
Dans le même temps, l’explicabilité peut aider les utilisateurs à comprendre, maintenir et utiliser l’IA, et à découvrir et comprendre de nouveaux concepts dans les domaines d’application de l’IA. L’explicabilité démontre également la faisabilité des résultats et indique aux utilisateurs quelle décision apportera le plus grand bénéfice.
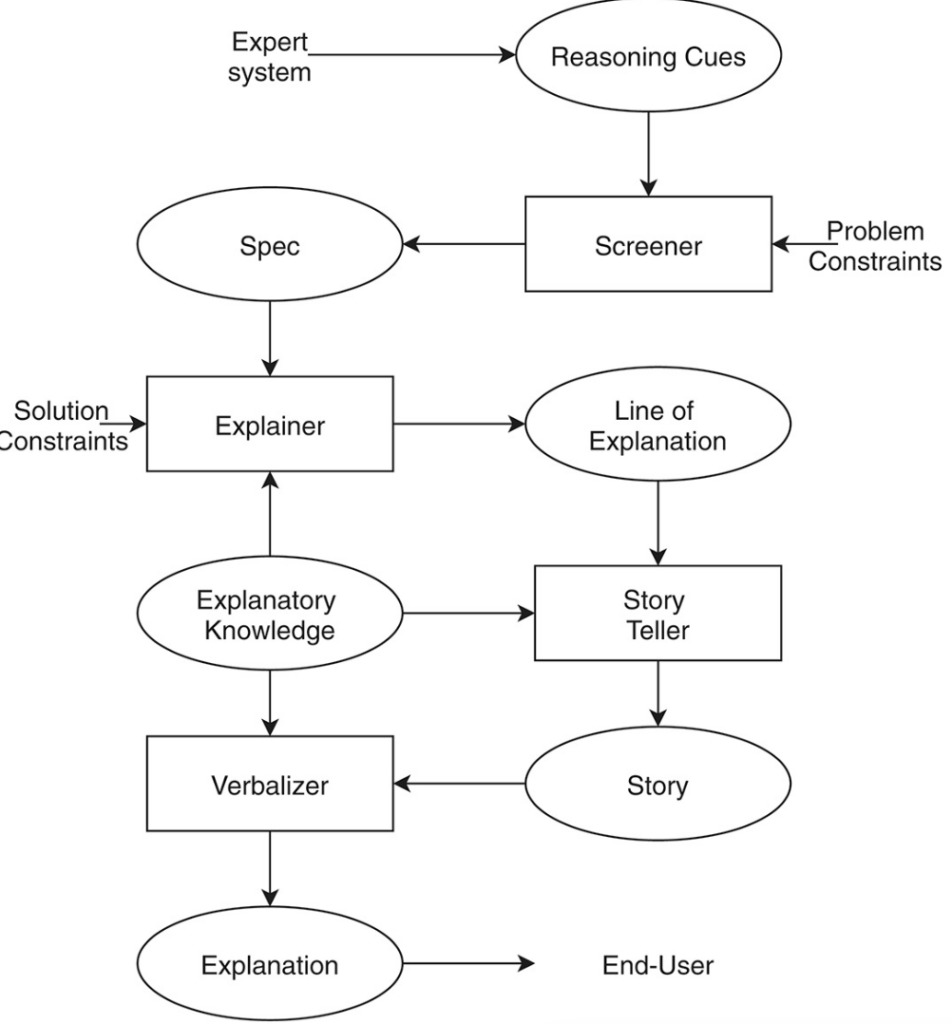
Figure 7 : Phase d'explication du processus de résolution de problèmes
Cependant,Les performances et l'interprétabilité des modèles constituent un problème majeur pour ScienceAISi le modèle présente de bonnes performances et une bonne robustesse entre les tests croisés, les caractéristiques profondes de grande dimension peuvent mieux fonctionner, mais elles n'ont aucune signification physique, ce que nous appelons souvent «L’interprétabilité de la recherche scientifique est généralement médiocre".
Au contraire, si des fonctionnalités bien expliquées sont utilisées, même si elles sont physiquement très interprétables, les performances réelles du modèle dépendront fortement des données, et les performances du modèle diminueront si l'ensemble de données est modifié.
Il n’existe pas encore de bonne façon d’unifier la contradiction entre les deux, mais dans cette étude, les chercheurs ont visualisé le processus de prise de décision de l’IA étape par étape., permettant aux utilisateurs de comprendre clairement les changements de score de divers résultats de prédiction à chaque étape, de comprendre le processus de prise de décision de l'IA et également de faciliter l'optimisation du modèle par les développeurs.
Avec le développement continu de l’IA explicable, la compréhension de l’IA par les gens deviendra plus approfondie et le processus de prise de décision de l’IA deviendra plus facile à comprendre.À l’avenir, l’interaction entre les humains et les machines continuera d’augmenter, le seuil d’interaction sera encore abaissé et l’IA sera utilisée dans davantage de scénarios, rendant la vie plus pratique et plus intelligente.
Liens de référence :
[1]http://www.chem.ucla.edu/~harding/IGOC/R/retrosynthesis.html
[2] https://zh.wikipedia.org/zh-cn/Spécification d'entrée linéaire moléculaire simplifiée
[3]https://wires.onlinelibrary.wiley.com/doi/10.1002/widm.1391








