Command Palette
Search for a command to run...
A French Team Successfully Predicted 2.39 Million Antiphage Proteins and Used a Deep Learning Model to Map Bacterial Antiviral immunity.

In the microscopic world, the "arms race" between bacteria and bacteriophages has never ceased. Bacteriophages typically outnumber bacteria by about 10, using bacteria as hosts for their own reproduction. Meanwhile, bacteria have developed highly diverse antiviral defense systems through long-term evolution. Currently, over 250 anti-phage systems have been experimentally validated, encompassing various mechanisms such as restriction-modification systems and CRISPR-Cas systems, and new systems are constantly being discovered. This phenomenon suggests that the complexity and diversity of bacterial defense systems may far exceed current understanding. However,Limited by traditional experimental methods and computational techniques, a large number of potential anti-phage mechanisms remain hidden in the bacterial genome and have not yet been systematically explored.
Existing research has noted certain common characteristics among known antiphage systems at the protein sequence and genome organization levels, such as the recurrence of characteristic domains and their enriched distribution in "defense islands" or prephage regions. These patterns suggest that:If these common patterns can be identified and utilized, it may be possible to systematically uncover unknown antiphage systems at the whole genome scale.
Based on this approach, researchers at the Pasteur Institute in France developed and fine-tuned three complementary deep learning models for large-scale prediction of phage resistance. The ALBERT_DF model relies solely on local genomic context for inference; ESM_DF utilizes a protein language model to parse amino acid sequences; and GeneCLR_DF integrates sequence information with genomic context. In a unified benchmark test,GeneCLR_DF performed best, achieving a precision of 991 TP3T and a recall of 921 TP3T.
Based on this high-precision model, the study further conducted pan-genome-scale predictions of antiphage systems. The results showed that in over 32,000 bacterial genomes, approximately 1.51 TP3T genes in a typical bacterial genome are involved in antiviral defense; more importantly, over 851 TP3T genes, representing a predicted defense-related protein family, had never before been associated with immune function. Ultimately,The model predicted approximately 2.39 million antiphage proteins, a large number of which belong to single-gene defense systems, and defined approximately 23,000 operon families based on gene co-occurrence relationships.The vast majority of these bacteria were previously unrelated to antiviral defense. These results collectively paint a systematic picture of bacterial antiviral immunity, revealing its scale and diversity far exceeding existing knowledge.
The related research findings, titled "Protein and genomic language models uncover the unexplored diversity of bacterial immunity," have been published in Science.
Research highlights:
* A total of 2.39 million antiphage proteins were predicted, of which 85% had never been associated with immune function before;
* In a typical bacterial genome, approximately 1.51 TP3T genes are specifically responsible for antiviral defense.
* Approximately 23,000 manipulator subfamilies were predicted, the vast majority of which were discovered for the first time;
* A large number of predicted defense proteins exist in the form of single-gene systems, challenging the conventional view that defense functions are usually accomplished by the collaboration of multiple genes.

Paper address:
https://www.science.org/doi/10.1126/science.adv8275
Follow our official WeChat account and reply "GeneCLR" in the background to get the full PDF.
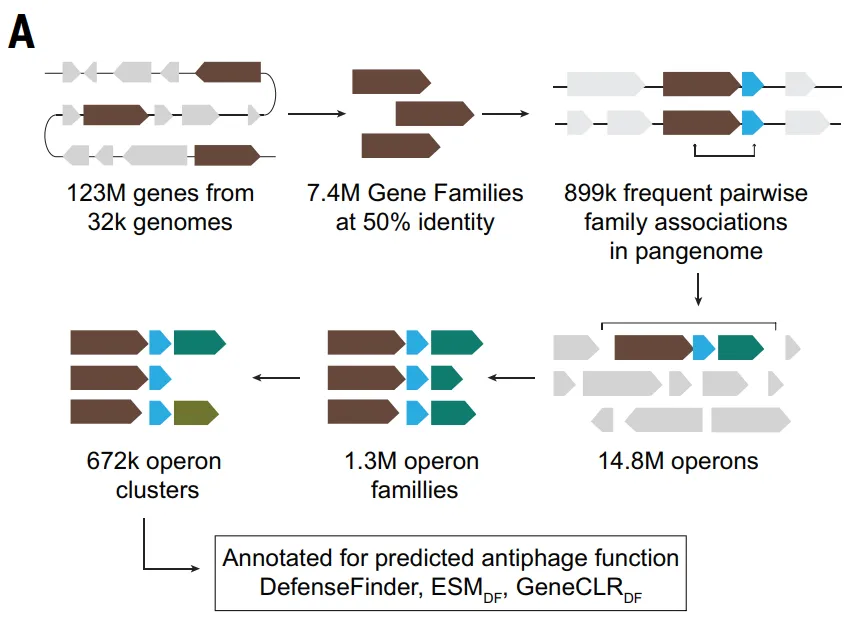
Dataset: Based on 123 million proteins and 32,000 genomes
This study first utilized the DefenseFinder and PadLoc tools,A systematic scan of 32,798 complete bacterial genomes in the RefSeq database was performed to quantitatively characterize known antiphage systems.Of the approximately 123 million proteins, DefenseFinder v1.3 identified 521,360, representing 0.41 TP3T, which belong to the antiphage system components, while PadLoc identified 805,357, representing 0.651 TP3T.
It is worth noting that many defense systems were initially discovered through genomic associations with known systems. These associations can be quantified at the protein family level using a “defense score,” which measures the frequency with which a particular protein family co-occurs with known defense proteins in the genome.
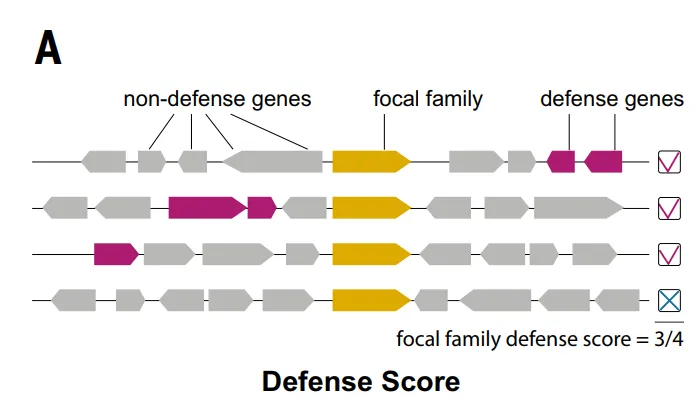
Based on the defense score method, as shown in the figure below.Researchers identified a total of 37,959 protein families (4.61% of TP3T) as candidate antiphage families.Subsequently, the study eliminated 7,799 families, such as integrases, that were associated with core biological functions or mobile genetic elements, ultimately resulting in 30,160 selected candidate families (accounting for 3.71 TP3T).
However, this method has obvious limitations:Firstly,It only applies to protein families containing more than five homologous sequences, thus excluding proteins of about 23%;Secondly,Some antiphage systems are not located in typical defense islands, and even if they have defensive functions, their defense scores may be low, thus causing them to be overlooked.
To overcome the above limitations and capture defense-related genomic signals more comprehensively,The study further constructed a dataset suitable for deep learning.Within the ALBERT_DF model framework, the study modeled the bacterial genome in a "linguistic" way: treating each protein family as a "word" and adjacent gene segments as a "sentence".
Because the complete dataset contains over 8 million different protein families, far exceeding the size of the vocabulary of traditional language models,The study limited the training scope to the phylum Actinobacteria, constructing a dataset containing 10,796 genomes.The genes were clustered into 4.2 million protein families, while the vocabulary was limited to the 524,288 most common families, thus covering approximately 891 TP3T proteins.
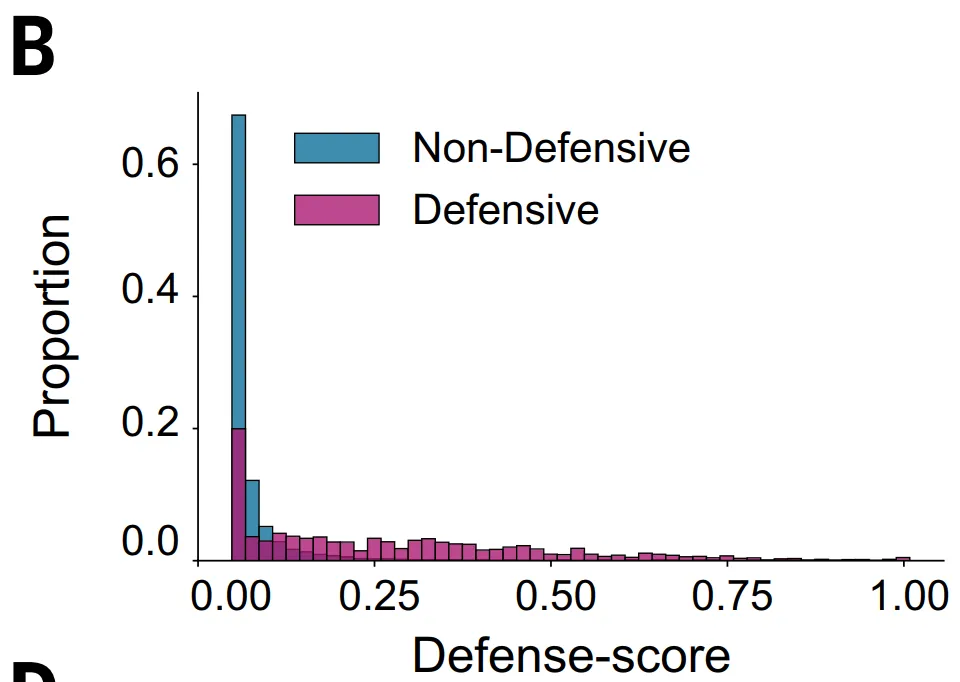
For the ESM_DF and GeneCLR_DF models, the study constructed the Gembase_DF dataset: as shown in the figure below, 521,360 antiphage proteins labeled with DefenseFinder were used as positive samples, 116 million highly conserved core genes present in more than 99% and 14 million non-defense mobile genetic element genes were used as negative samples, and the remaining proteins were retained as unlabeled candidates.
To avoid information leakage between training, validation, and testing, the study grouped all proteins of the same defense system into the same data fold and used MMseqs2 to remove residual homology across data folds, ensuring the rigor of model evaluation.
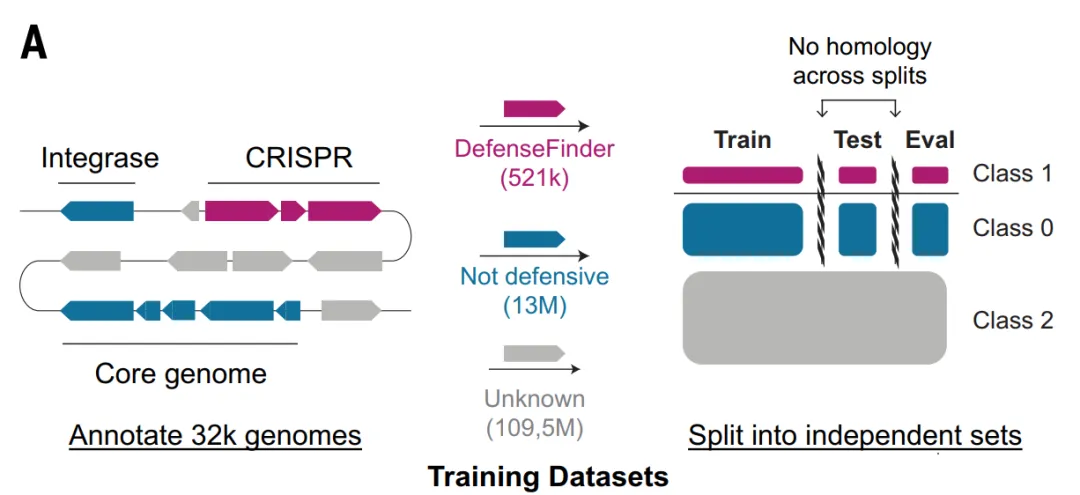
Model architecture: A three-layer deep learning model that progresses step by step.
To overcome the limitations of traditional "defense score" methods, the research team constructed a complementary and progressive deep learning framework, targeting three objectives: discovery of unknown systems, pan-genome-scale mining, and high-precision integrated prediction.Specifically, this includes ALBERT_DF based on genomic context, ESM_DF based on protein sequence, and GeneCLR_DF which integrates sequence and contextual information.
Among them, ALBERT_DF focuses on learning functional signals from gene "neighborhood relationships" and has the ability to discover novel defense systems; ESM_DF directly uses amino acid sequence modeling and has good cross-sequence generalization ability; while GeneCLR_DF integrates the two types of information in a unified framework and achieves a better balance between recognition accuracy and prediction coverage.
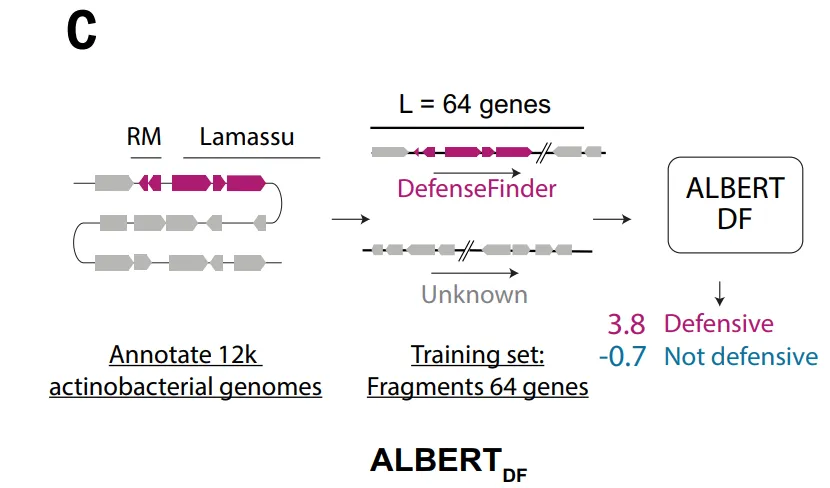
The ALBERT_DF model is based on a key observation: antiphage systems tend to be clustered throughout the genome, with stable organizational patterns existing within and between neighboring genes. Based on this characteristic,This study introduces the ALBERT architecture from natural language processing into genome modeling.Treating protein families as "words" and gene sequences as "syntactic structures," we learn local context by predicting masked genes.
Unlike traditional sequence-similarity-based methods, this modeling approach directly utilizes genomic organization information, thus holding greater potential for identifying novel defense mechanisms that lack homology with known systems. However, due to its reliance on discretized "lexical" representations, this type of method has inherent limitations when expanding across species.
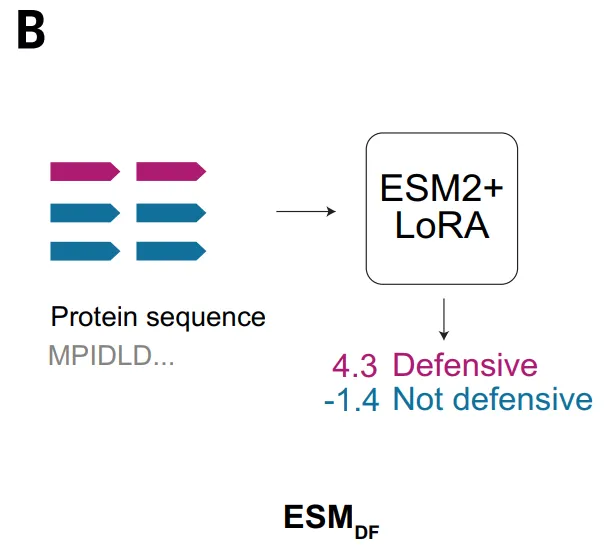
The ESM_DF model, on the other hand, takes a different approach, acting directly on the protein amino acid sequence.This model learns the co-variations between residues and long-range sequence relationships through large-scale pre-training.This allows for the extraction of functional signals without relying on artificial features. After fine-tuning, ESM_DF can score any protein to determine whether it participates in anti-phage defense. This approach significantly improves the applicability of the method, enabling it to operate at a pan-genome scale. However, at the same time, ESM_DF's discriminative ability still depends to some extent on sequence similarity, thus it is better at identifying distant variants of known defense systems, and its ability to identify novel domains lacking homology is relatively limited.
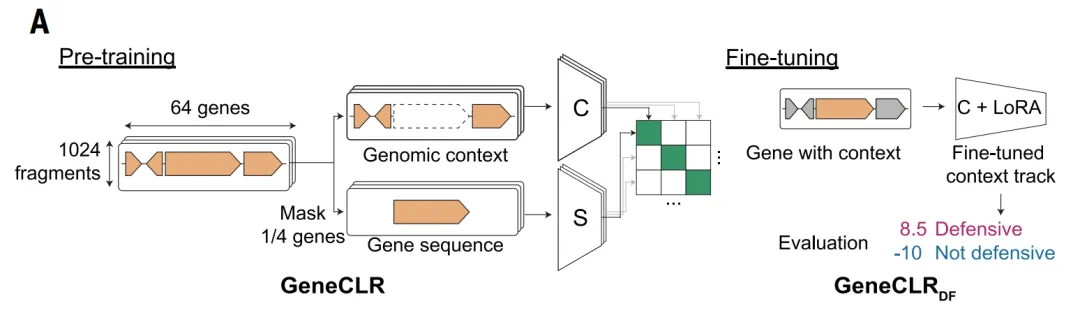
Based on this, the GeneCLR_DF model was proposed to integrate sequence and genomic context information.This model employs a contrastive learning framework, simultaneously learning two representations for each gene:One type of representation comes from the protein sequence, and the other comes from its genomic neighborhood. By training the model, it is determined whether these two representations correspond to the same gene, thereby aligning the two types of information in the representation space.
This design offers a key advantage: when certain genes lack homology at the sequence level, their typical genomic context can still provide identification clues; conversely, when contextual information is atypical, sequence features can still support discrimination. Through this complementary mechanism,GeneCLR balances the ability to discover novel systems with the scalability for large-scale applications in subsequent predictions.
Overall, these three types of models form a clear technical path: from context-based local pattern learning to sequence-based global generalization, and then to unified modeling of multi-source information. This hierarchical design not only avoids the limitations of a single method but also provides a more universal technical framework for systematically exploring unknown antiphage mechanisms.
Achieve 991 TP3T precision and 921 TP3T recall.
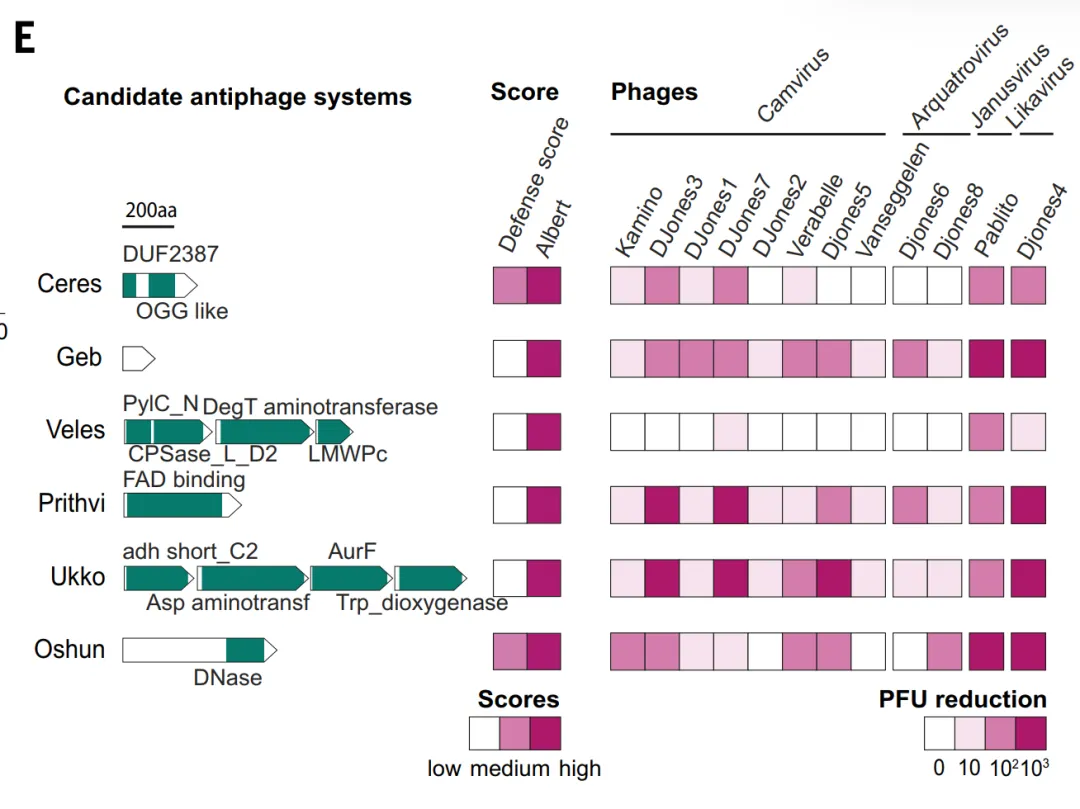
In the experimental validation, the study first evaluated the predictive power of ALBERT_DF.The model predicted a total of 1,930 candidate antiphage protein families, of which approximately 331 TP3T overlapped with the results of the defense score method.Researchers further selected 10 candidate systems that lacked both defense score support and known homology, expressed them in *Streptomyces whiteus*, and challenged them with 12 phages. Six of these systems exhibited robust protection, reducing plaque-forming units by more than 100-fold. These systems (such as Ceres and Geb) contain metabolic enzymes and small proteins with unknown functions, exceeding the scope of classical defense domains, demonstrating that genomic context-based methods can discover novel defense mechanisms that are difficult to identify using traditional methods.
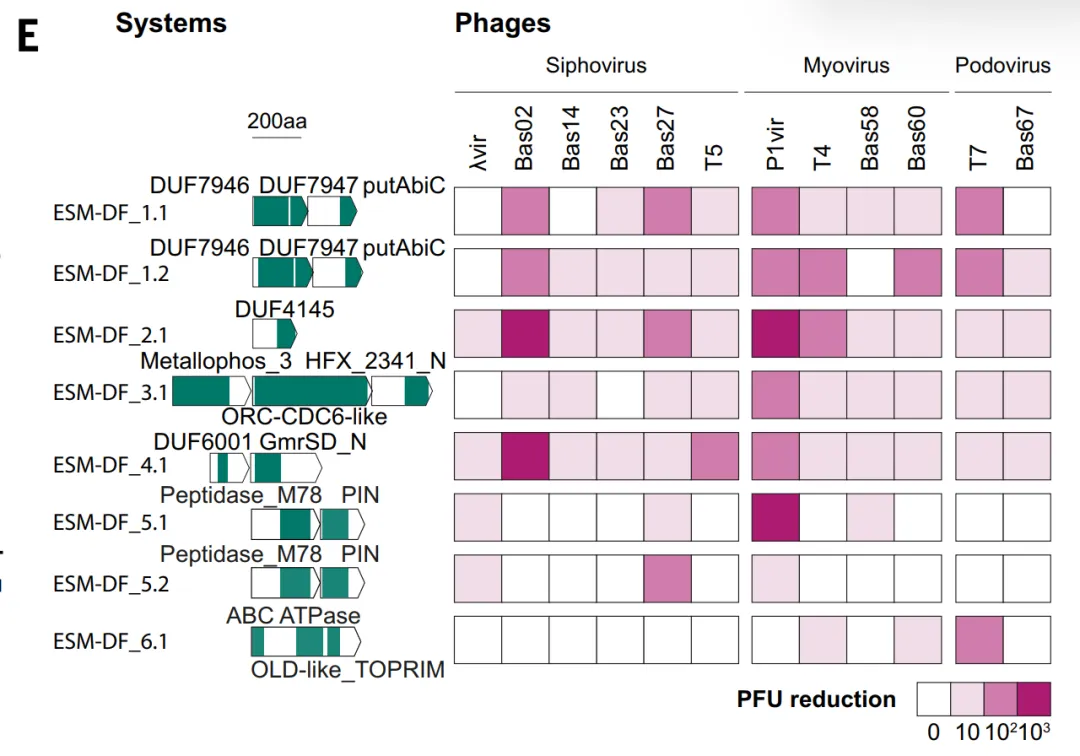
In the validation of ESM_DF, the study tested a group of high-scoring candidates in E. coli, of which six systems demonstrated antiphage capabilities, including ESM_DF, which is resistant to multiple types of bacteriophages. These systems included variants of known defense domains as well as domains not previously associated with antiphage function, such as DUF7946.This indicates that ESM not only relies on sequence homology but can also identify a wider range of functional features, but overall it still tends to be an extension of known systems.
GeneCLR_DF performed best in the system evaluation. On the test set,Its prediction scores can clearly distinguish between defensive and non-defensive proteins.In evolutionary analysis, it consistently assigned high scores to key defense branches such as retrotranscriptors, CBASS, and Thoeris, while ESM-650M_DF could only partially identify them.
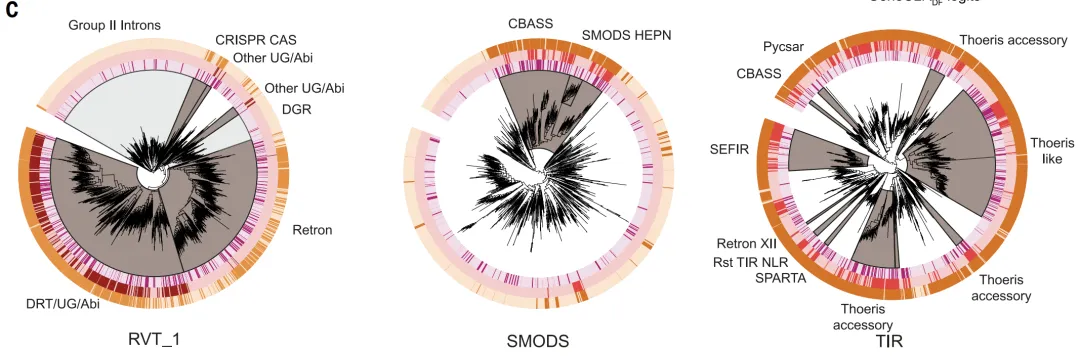
In different genomic contexts (defense islands, integrons, prephage regions),GeneCLR_DF can accurately locate the defense module.Quantitative results showed that at a threshold of −0.74, GeneCLR_DF achieved a precision of 991 TP3T and a recall of 92.41 TP3T; at the same precision, ESM_DF recalled only 581 TP3T. With a false detection rate of 11 TP3T, GeneCLR_DF retrieved 941 TP3T in known defense families, significantly higher than ESM-650MDF (351 TP3T) and the defense fraction method (51 TP3T), and identified only 561 TP3T families; it also recovered 751 TP3T from the 110 newly added systems. Of the 615,672 candidate protein families, 931 TP3T were detected only by GeneCLR_DF.
At the operon level, further analysis based on collinear clustering revealed that a large number of defense structures remain unknown: the predicted protein family of 85% was identified only by ESM_DF and GeneCLR_DF, while the operon family of 45% and the operon cluster of 52.7% previously lacked functional annotations. Evolutionary analysis also revealed that...The median proportion of defense genes in the bacterial genome increased from 0.46% to 1.53%.Furthermore, a large number of systems are enriched in mobile genetic elements, with 23.5% located within the MGE boundary and satellite elements of 47.1% predicted to encode defense capabilities.
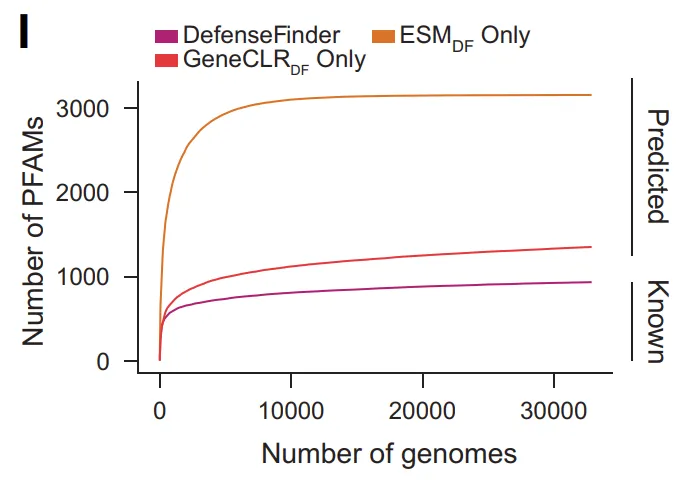
At the molecular diversity level, GeneCLR_DF expanded the number of defense-related Pfam families from 934 to 3,154 (approximately 15% of all Pfams). Simultaneously, over 400,000 predicted protein families lacked any Pfam annotations, with fewer than 5% appearing in DefenseFinder; over 3,500 operon families consisted entirely of proteins without known domains. These results indicate that...Much of the molecular space of antiphage defense has not yet been systematically characterized.
Deep learning drives a leap in the efficiency of antiphage defense discovery
Deep learning-based antiphage system prediction frameworks and the bacterial antiviral immune atlases constructed from them are opening up a more scalable research path in this field: shifting from "point-based breakthroughs" relying on individual case discoveries to "systematic mining" based on pattern recognition. This change not only improves the efficiency of discovering novel defense mechanisms but also brings academic research and industrial applications closer together.
In academia, this approach has been rapidly expanded. Multiple research institutions have begun combining machine learning with genomic analysis to attempt to identify phage-resistant systems on a larger scale. For example,The DefensePredictor model, developed by a team at MIT,By drawing on the modeling logic of protein language models and integrating gene sequence and genomic context information, a highly sensitive identification of antiphage proteins was achieved. The model was trained on approximately 17,000 prokaryotic reference genomes and identified approximately 821 TP3T novel defense systems in independent tests, further validating the feasibility of "pattern-based discovery of unknown functions."
Paper Title: DefensePredictor: A machine learning model to discover prokaryotic immune systems
Paper link:
https://www.science.org/doi/10.1126/science.adv7924
In the industry, related technologies are also being rapidly implemented. With the growing severity of antibiotic resistance, bacteriophages and their derivative technologies are regaining importance, becoming a crucial direction for replacing or supplementing traditional antibiotics. Locus Biosciences, a clinical-stage company, has built a platform based on engineered bacteriophages, combining machine learning and synthetic biology to develop LBP-EC01, a candidate therapy for multidrug-resistant E. coli, thus advancing the precision and controllability of phage therapy.
Meanwhile, Micreos takes a more application-oriented approach, focusing on the industrialization of bacteriophages and endosomalins. Its product Listex has been used in food processing to inhibit Listeria contamination and has received regulatory approvals in multiple countries; Staph Efekt utilizes the specific bactericidal capabilities of endosomalins in skin care. This approach emphasizes "functional implementation"—transforming antiphage mechanisms into concrete, usable products, rather than simply remaining at the laboratory level.
Overall, from algorithmic models to experimental verification and then to industrial applications, antiphage research is gradually forming a more complete chain. It is foreseeable that with the accumulation of more data and the iteration of models, this path, starting with computation, verifying through experiments, and guided by applications, will continue to drive a deeper understanding of bacterial immune systems and more effectively translate these findings into real-world solutions.
Reference Links:
https://mp.weixin.qq.com/s/usrVEOeBD5gphhslZahLCA
https://mp.weixin.qq.com/s/Pxlh69TXSr8ffAp_ul3URw








