Command Palette
Search for a command to run...
Breaking Through the Limitations of Traditional Multimodal Integration! MIT Proposes the APOLLO Framework, Achieving a Clear Separation of cell-shared and cell-specific information.
In single-cell biology research, the rapid development of measurement technologies is constantly expanding the boundaries of scientific exploration. Breakthroughs in areas such as multiplex imaging, single-cell transcriptome sequencing (scRNA-seq), chromatin open sequencing (scATAC-seq), and protein abundance detection have enabled researchers to conduct panoramic observations of single cells from multiple dimensions, including transcriptional regulation, chromatin state, protein expression, and morphological structure. These multimodal data interpret the code of life from different levels, and their complementary integration provides an unprecedented opportunity to reveal cellular heterogeneity and explore disease mechanisms.
However, current analytical methods still have significant limitations when dealing with such high-throughput data.Mainstream strategies often involve analyzing each mode separately and then comparing them, which is not only inefficient but also makes it difficult to capture the deep correlations between modes.Another type of approach integrates multimodal data into the same latent space through representation learning, but often confuses shared information with modality-specific information, obscuring the unique contribution of each dimension to cell function.
This problem is particularly prominent in the integrated analysis of paired scATAC-seq and scRNA-seq data.Traditional methods often coarsely granulate chromatin accessibility to the gene level for comparison with gene expression. While this simplifies the problem, it may discard fine structural information at the chromatin level and is only applicable to data types with relatively uniform characteristics. More complex integration methods, such as linear models and generative adversarial networks, either struggle to adapt to unstructured data like imaging or perform poorly in separating shared and specific information, failing to meet the growing demand for multimodal data analysis in large biobanks.
Therefore, with the continuous evolution of single-cell technology and the rapid growth of data scale, how to efficiently and automatically integrate multimodal data while clearly decoupling shared information from modality-specific information has become a core challenge facing single-cell biology.
To address this challenge, a joint research team from MIT and ETH Zurich proposed a general deep learning computing framework called APOLLO (Autoencoder with a Partially Overlapping Latent space learned through Latent Optimization).This framework provides a feasible technical path for a more comprehensive and accurate analysis of cell states and their regulatory logic by explicitly modeling shared information and modality-specific information.
The related research findings, titled "Partially shared multi-modal embedding learns holistic representation of cell state," have been published in Nature Computational Science.
Research highlights:
* This research proposes APOLLO, a general deep learning framework that can automatically and explicitly decouple "shared information" from "modality-specific information" in multimodal data.
* APOLLO learns a partially overlapping latent space by equipping each modality with an autoencoder and employing a two-step training strategy, thereby effectively identifying and distinguishing biological signals commonly captured across multiple modalities.
* APOLLO can reveal the association between differences in protein subcellular localization and the morphology of different cell compartments, thus extending the analysis from purely omics data to the field of spatial morphology.

Paper address:
https://www.nature.com/articles/s43588-025-00948-w
Follow our official WeChat account and reply "APOLLO" in the background to get the full PDF.
Dataset: Comprehensive validation covering sequencing and imaging
To comprehensively evaluate the performance of the APOLLO framework, the study used multiple publicly available multimodal single-cell datasets, covering both sequencing and imaging technologies.
Regarding sequencing data,Researchers first used paired single-cell transcriptome (scRNA-seq) and chromatin accessibility (scATAC-seq) data measured by SHARE-seq technology to verify whether APOLLO could automatically identify and distinguish gene activity captured by both transcriptome and chromatin accessibility, as well as gene activity captured by only a single modality of either.
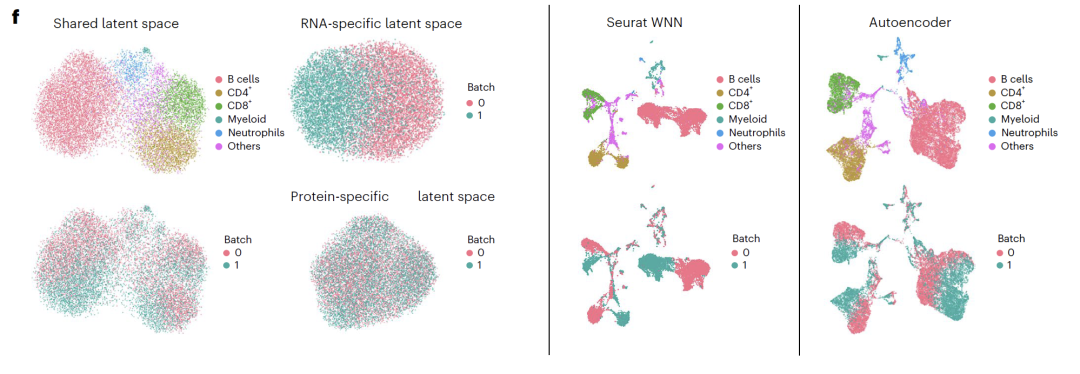
Secondly, the researchers used paired scRNA-seq and cell surface protein abundance data obtained by CITE-seq to further test the applicability of the model to sequencing data.The CITE-seq dataset is derived from mouse spleens and lymph nodes, and includes two groups of wild-type mouse samples that underwent independent experimental treatment.It can not only be used to assess cell type discrimination ability, but also reveals the experimental batch effect caused by different mouse sources.
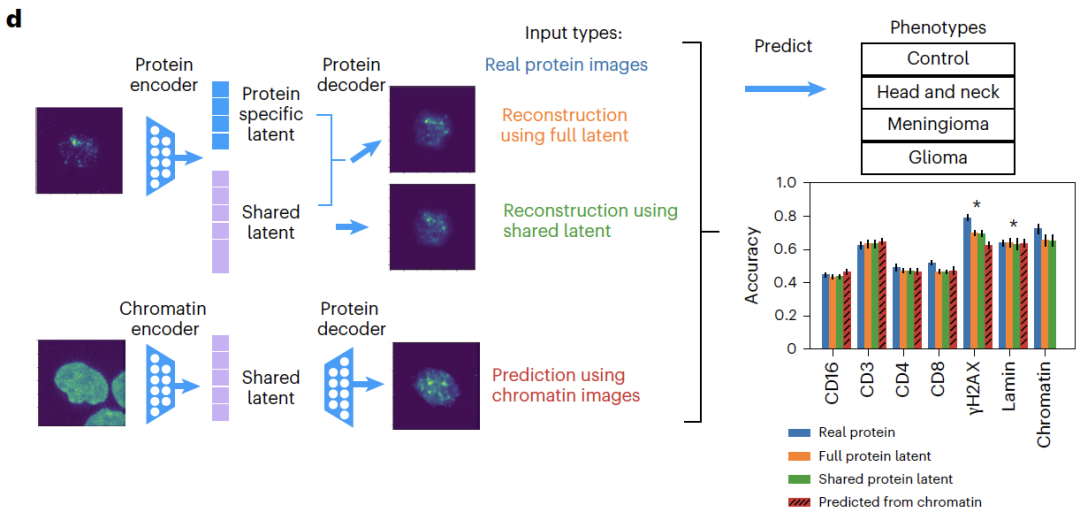
Regarding imaging data,Researchers introduced a multiplex imaging dataset of human peripheral blood mononuclear cells (PBMCs), encompassing 32,345 cells from 40 patients, categorized into four diagnostic categories: healthy, meningioma, glioma, and head and neck tumors. Two sets of imaging data were collected from each patient based on different antibody combinations: one set used DAPI to label chromatin and combined with CD4, CD8, and CD16 antibody staining; the other set also used DAPI staining but combined with lamin, CD3, and γH2AX antibody staining.
Tests using this dataset revealed that,APOLLO can identify cellular state information shared between the two modalities in chromatin structure and protein localization, as well as morphological features captured by only a single modality.In addition, by combining additional cell staining markers such as microtubules and endoplasmic reticulum, the study also used multiple imaging data from the Human Protein Atlas (HPA) to demonstrate that APOLLO can be used to reveal the association between differences in protein subcellular localization and the morphology of different cell compartments.
APOLLO Model: An autoencoder employing a latent optimization strategy
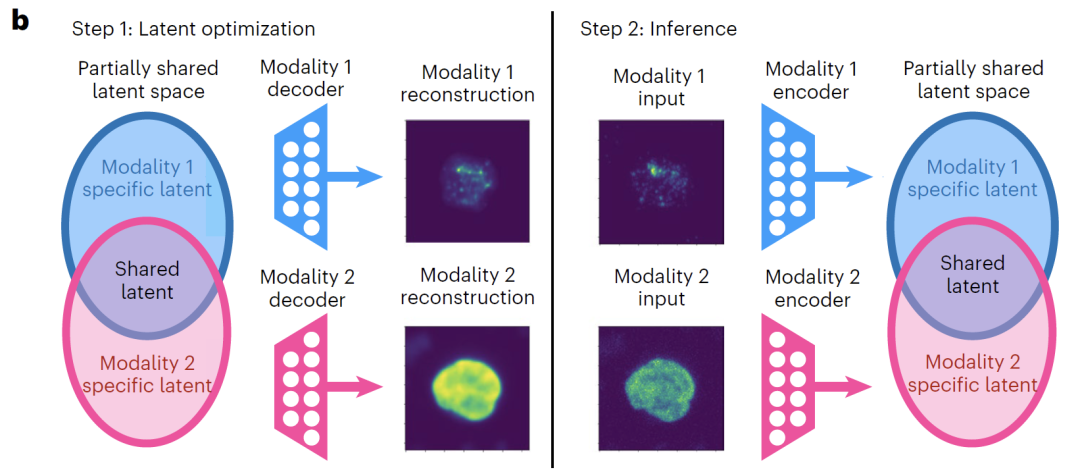
To address the common problem in existing multimodal integration methods that confuse shared information with modality-specific information, this study proposes the APOLLO framework. This framework employs latent optimization to learn an autoencoder in a partially overlapping latent space, aiming to automatically learn and effectively decouple shared and modal-specific information across multiple modalities. Unlike conventional autoencoders that uniformly align all latent dimensions,APOLLO performs cross-modal alignment only on some of the potential dimensions, reserving the remaining dimensions for information specific to each modality, thus achieving a clear separation between shared and specific information in model design.
In terms of model architecture,APOLLO is equipped with an autoencoder for each data modality and can introduce additional decoders as needed for the task.The encoder and decoder employ neural network structures adapted to specific modalities; for example, convolutional networks are used for imaging data, while fully connected networks are used for gene expression data, to fully capture the data characteristics of each modality. The latent space is explicitly divided into two parts: shared latent features and modality-specific latent features. The dimension of the shared latent space is usually set much larger than that of the modality-specific space to ensure sufficient representation of cross-modal shared information.
As shown in the figure below, the training process of APOLLO consists of two steps:The first step focuses on training the decoders for each modality, while simultaneously updating the latent space.The core objective is to enable the decoder to accurately reconstruct the input data from the latent space. If the task requires strengthening the shared information representation and achieving cross-modal prediction, two additional decoders are introduced to map the shared latent space to each modality respectively, and training is completed by minimizing the reconstruction loss.
The second step is to train the modality-specific encoder.Each data mode is mapped to its corresponding latent space. By minimizing the mean square error, the embedding in the latent space of samples not involved in the training is inferred, thereby ensuring that the model has good generalization ability.
For model validation, the study first tested APOLLO's decoupling performance on five simulated datasets with known real underlying structures.The results show that the model can maintain stable performance regardless of the dependency between shared and specific latent features.Further validation on real data shows that APOLLO's explicit learning of partial information sharing can not only decouple multimodal information, but also achieve accurate cross-modal predictions, such as predicting undetected proteins from chromatin imaging.
Overall, APOLLO effectively decouples and interprets shared and modality-specific information in multimodal datasets by learning partially shared latent spaces, providing a general framework for uncovering biological mechanisms.
Beyond traditional multimodal integration frameworks, a more comprehensive understanding of cellular states.
To comprehensively evaluate the universality and core advantages of the APOLLO model, a series of experiments were designed around five directions: pair sequencing data integration, chromatin and protein imaging integration, cross-modal prediction, morphological feature recognition, and protein subcellular localization exploration.
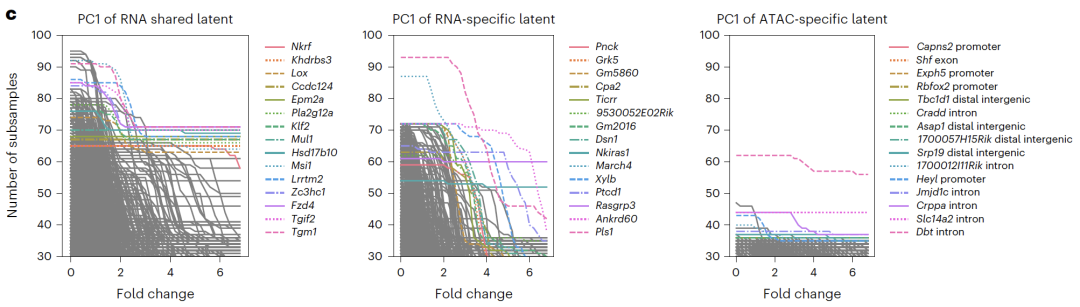
In paired sequencing data integrationSHARE-seq experiments showed that adding a modality-specific space to the shared space can significantly improve the accuracy of cell type classification, proving that the specific space can capture biological information not included in the shared space.
Potential spatial interpretation revealed that RNA-specific spaces were enriched with cell cycle-related genes, ATAC-specific spaces were enriched with open chromatin regions related to transcriptional regulation, and shared spaces were enriched with known transcription factors and regulatory pathways, validating the biological significance of the decoupling results. In CITE-seq experiments,APOLLO successfully separated cell type and batch effects into a shared space and an RNA-specific space.Existing integration methods cannot achieve this kind of decoupling, highlighting the unique advantages of the model in integrating sequencing data.
Regarding the imaging data,APOLLO can accurately reconstruct cell images from patients who did not participate in the training.In the cross-modal task of predicting undetected proteins from chromatin, APOLLO significantly outperforms traditional image inpainting methods;Downstream phenotypic classification showed that the classification accuracy based on predicted protein imaging was similar to that of actual imaging, with CD3 protein showing the best prediction performance, confirming that the prediction results can be effectively used for biological discovery.
In morphological feature recognition tasks,The shared space primarily captures chromatin morphological features (such as nuclear area and heterochromatin volume), while protein-specific features, such as γH2AX focal count, exist only in their corresponding specific spaces. Feature ablation experiments showed that removing this feature significantly reduced phenotypic classification accuracy, further validating the accuracy of the decoupling.

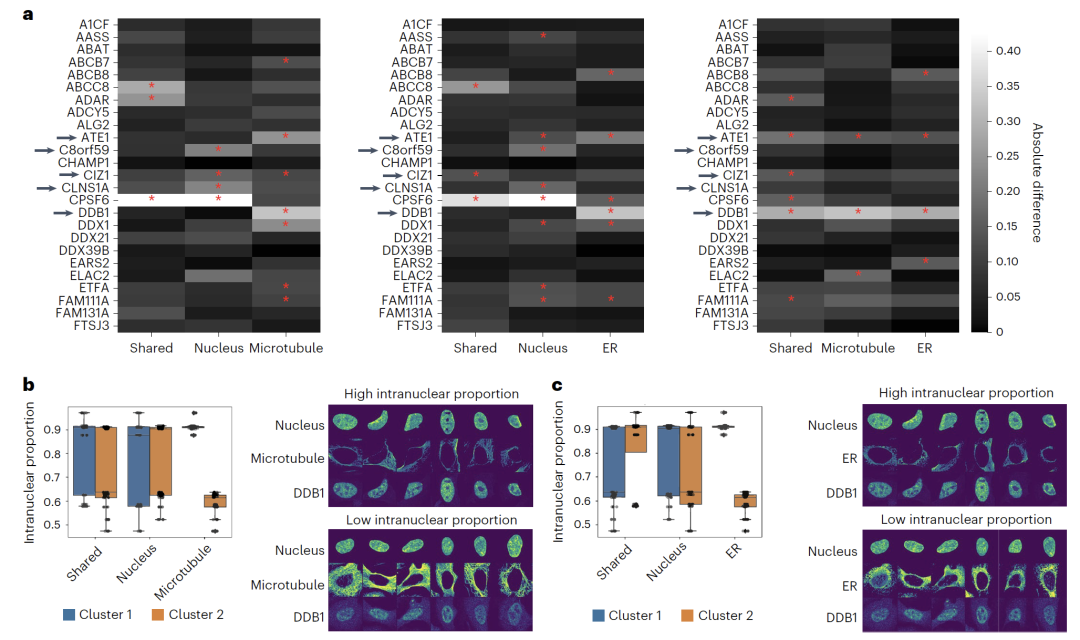
In the study of protein subcellular localizationApplying APOLLO to U2OS cell imaging data revealed that differences in protein localization within the nuclear cavity can be captured by characteristics of different cellular compartments. For example, the nuclear localization of DDB1 is associated with endoplasmic reticulum and microtubule morphology, while CLNS1A is associated only with nuclear morphology. This result indicates that...The model can be extended to various imaging combinations, providing a new perspective for understanding the relationship between protein localization and cell morphology.
Implementation of single-cell multimodal data integration
The integration of single-cell multimodal data is becoming a core technological direction for analyzing cellular heterogeneity, revealing disease mechanisms, and promoting the development of precision medicine, and has attracted widespread attention from the global academic community.
For example, the scMTR-seq technology developed by Peter Rugg-Gunn's team at the Babraham Institute at the University of Cambridge...For the first time, simultaneous capture of six histone modifications and the entire transcriptome was achieved at the single-cell level.They overcame a technical bottleneck in the field of epigenetics research that had lasted for a decade.
Paper Title:
Combinatorial profiling of multiple histone modifications and transcriptome in single cells using scMTR-seq
Paper link:
https://www.science.org/doi/10.1126/sciadv.adu3308
The CellFuse framework proposed by the Stanford University research team constructs a shared embedding space based on supervised contrastive learning, and is specifically designed for multimodal integration scenarios with limited feature overlap.It can achieve accurate cell type prediction and seamless integration across modalities and experimental conditions.Tests on multiple datasets, including healthy PBMCs, bone marrow, CAR-T therapy for lymphoma, and tumor tissue, demonstrate that the framework outperforms existing methods in both integration quality and operational efficiency.
Paper Title:
CellFuse Enables Multi-modal Integration of Single-cell and Spatial Proteomics data
Paper link:
Meanwhile, leading global biotechnology and healthcare companies are accelerating the deployment of single-cell multimodal data integration technology, focusing on core scenarios such as clinical translation, drug development, and precision medicine, to drive the transformation of cutting-edge research into practical applications. German company BioNTech has applied this technology to tumor immunotherapy and personalized vaccine development. By integrating single-cell RNA sequencing, protein expression profiling, and spatial transcriptomics data, they precisely analyze cellular heterogeneity in the tumor microenvironment, identify key immune cell subtypes and related biomarkers, providing core data support for the design and optimization of personalized tumor vaccines, and significantly improving vaccine targeting and efficacy.
It is foreseeable that, driven by continuous breakthroughs in multimodal integration technology, the decoding of life at the single-cell level will eventually move from vision to reality, injecting stronger momentum into the future of precision medicine.