Command Palette
Search for a command to run...
ICLR 2026 | NVIDIA/Oxford University and Others Propose an atomic-level Protein Binder Generation Method With state-of-the-art (SOTA) performance.

In the field of computational biology,Designing proteins that can bind precisely to specific targets is one of the most critical and challenging problems.It is not only directly related to key areas such as drug development, biotherapy and enzyme engineering, but also determines the upper limit of human efficiency in the intervention of complex diseases and biomanufacturing.
From a molecular perspective, the binding of a protein to a target is essentially a three-dimensional structural issue:The amino acid composition, spatial conformation, and intermolecular interactions of the interface together shape the affinity and specificity of the binding.Therefore, almost all binder design methods ultimately return to the core variable of "structure," using structure analysis or prediction to guide molecular construction.
In recent years, the introduction of machine learning has been reshaping this paradigm. With breakthroughs in structure prediction and structure generation models, research is gradually moving away from its strong dependence on experimental structures, shifting from "analyzing structures" to "generating structures," making it possible to design binders from scratch, and significantly reducing R&D costs and time.
However, in terms of methodology, current AI-driven binder design still exhibits a clear divergence:One type is generative methods, represented by RFDiffusion.It relies on large-scale training to directly generate candidate structures, but lacks the ability to flexibly adjust during the inference stage;Another type is the illusionary method, represented by BindCraft.Gradient optimization through scoring of structure predictors offers flexibility but lacks generative priors, making it difficult to explore entirely new structural spaces. This "separation of generation and optimization" contrasts with the unified paradigm of "pre-trained models + inference-time computational extensions" already established in the fields of natural language and image processing.
It is in this context thatA joint research team from NVIDIA, Oxford University, the Quebec Artificial Intelligence Institute, and other institutions proposed the Proteina-Complexa (hereinafter referred to as Complexa) framework.Aimed at bridging the gap between generative and illusionary methods, this approach unifies the basic generative model and inference-time optimization mechanisms within a single system. It is based on Teddymer pre-training.Complexa enables state-of-the-art de novo binding agent design without requiring additional sequence redesign steps.By adapting the test-time scaling technique from the diffusion model to this framework, generation and optimization are directly unified, thus outperforming traditional illusion-based methods in terms of performance.
The related research findings, titled "Scaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute", have been accepted for ICLR 2026.
Research highlights:
* This study proposes Complexa, which extends La-Proteina to binder design, utilizes Teddymer, and achieves efficient inference-time optimization accelerated by generative priors.
* Achieved state-of-the-art computer simulation success rates in protein and small molecule target and enzyme design benchmarks without requiring sequence redesign.
Paper address:
https://openreview.net/forum?id=qmCpJtFZra
Follow our official WeChat account and reply "Complexa" in the background to get the full PDF.
Datasets: From "Single-unit Enrichment" to "Complex Reconstruction"
A fundamental limitation of binder generation models lies in the data. Ideally, the model requires a large amount of "binding agent-target complex" data for training, but in reality, this type of data mainly comes from experimentally analyzed protein databases (PDBs), which are limited in scale and high-quality samples are even scarcer; while the larger AlphaFold database (AFDB) provides a massive amount of protein structures, almost all of them are monomeric structures, lacking complex information.This structural gap of "abundant individual components and scarce complex components" directly limits the model's ability to be trained on a large scale.
The key breakthrough of this study comes from a renewed understanding of the internal structure of AFDB. Most proteins in AFDB are multi-domain proteins, and the domain segmentation tool TED provides them with fine annotation. Further analysis revealed that the interactions between different domains within the same protein exhibit statistically similar characteristics to those in multi-stranded complexes. This observation leads to an important shift:Monomer structures are not "useless data" in themselves, but can be reinterpreted as a potential source of data for complex structures.
Based on this insight, the study proposes a method for "artificial multimer construction".By breaking down multi-domain proteins into independent domains and treating them as different chains, a complex-like structure can be constructed within the monomer.The specific process starts with AFDB50, screening proteins with TED annotations, then splitting them into "pseudo-multimers," further extracting dimers and filtering them based on spatial proximity, while retaining samples with complete annotations. Finally, after clustering to remove redundancy, approximately 3.5 million dimer clusters are obtained. This dataset is named Teddymer, and its essence is not simply expanding the data scale, but rather transforming "monomer advantage" into "complex supply" through structural reorganization.
During the training process, as shown in the figure below, the study did not rely on a single data source, but integrated AFDB monomer data, Teddymer construction data, PDB experimental complex data, and PLINDER protein-ligand data, enabling the model to establish a unified representation among monomer structure, complex structure, and small molecule interactions, thereby taking into account both generative and generalization capabilities.
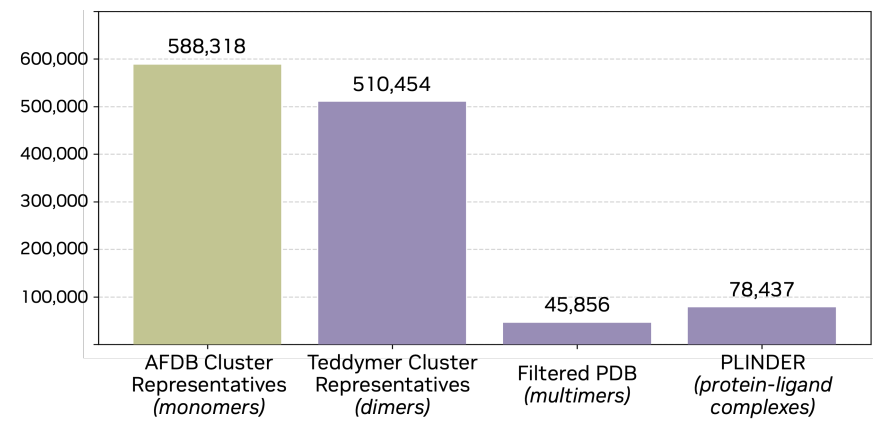
Complexa: A full-atomic framework for protein binding agent generation
In terms of model design, the core change in Complexa is not merely "stronger generative ability," but rather a shift in the generative target from "complete protein structures" to "binding agents at specific interfaces." Built upon La-Proteína, its advantage lies in its ability to generate at the entire atom level.At the same time, based on the efficient Transformer architecture, it avoids modules with high computational costs in traditional structural models, giving it good scalability in large-scale sampling scenarios.
Building upon this, the study introduces a generation mechanism conditioned on target points and interface hotspots. This mechanism prevents the model from generating complete complexes, instead generating only the binder portion, and explicitly relies on target information during the generation process. Specifically,The flow matching model is responsible for generating structures under conditional constraints, while the autoencoder is only used for encoding and decoding of monomer binders, thereby reducing modeling complexity while maintaining expressive power.
To enable the model to effectively understand target information, a systematic design for the input representation was implemented. Protein targets were encoded using the Atom37 method, with residue-level 3D coordinates, amino acid types, and interface hotspot information uniformly input into the model. Hotspots indicate potential binding regions. During training, these hotspots were extracted from real interfaces, while during inference, they were used as priors or obtained through preprocessing. For small molecule targets, the model encoded type, charge, and spatial coordinates at the atomic level, and these were input together with the binding agent representation into the Transformer for joint modeling.
Regarding training objectives,A key improvement is the introduction of random global translation noise into the binder coordinates, thereby forcing the model to learn the spatial localization ability of molecules.This is not important in monomer generation, but it is a key capability that determines the quality of generation for tasks that require precise placement of the binder at the target interface. The entire training process adopts a phased strategy, progressing step by step from monomer modeling to general structure generation, and then to binder-specific training. At the same time, overfitting is controlled through LoRA, and the monomer-level autoencoder is reused throughout to keep the architecture simple.
In the inference stage,Complexa further introduces a "test-time computation expansion" mechanism, which combines the generation process with search optimization.By increasing the number of samples, introducing bundle search, or Monte Carlo tree search, the model can continuously improve the quality of its generation under a larger computational budget. This design allows the model's capabilities to expand dynamically during inference, rather than being entirely limited to the training phase.
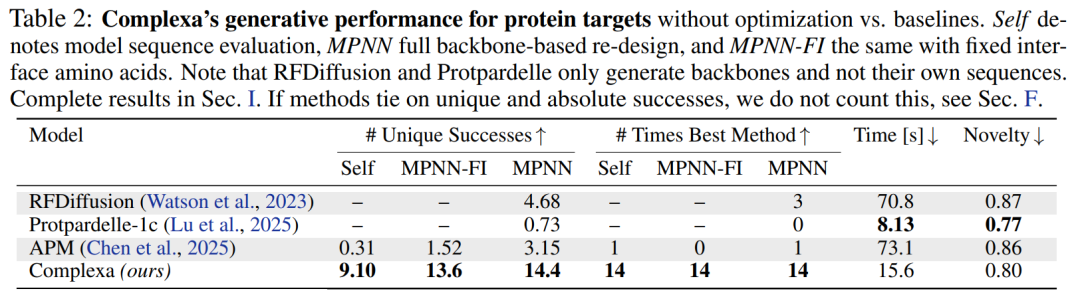
Higher success rate, faster speed, and stronger scalability
To validate the model's capabilities, this study designed a series of experiments ranging from simple to complex. The core question is: Does Complexa not only outperform in basic performance, but also continue to improve its performance as computing resources increase?
In terms of basic generation capabilities,Whether targeting proteins or small molecules, Complexa significantly outperforms existing methods, boasting higher success rates and faster sampling speeds.At the same time, the novelty of the generated structures is significantly improved. More importantly, the model can directly output high-quality sequences without relying on tools such as ProteinMPNN for secondary design, thus simplifying the overall process.
In terms of structural controllability,The study introduces conditional labels, enabling the model to explicitly control the type of generated structure.For example, choosing between α-helices and β-folds can effectively alleviate the problem of the single structure of previous generative models and significantly improve structural diversity.
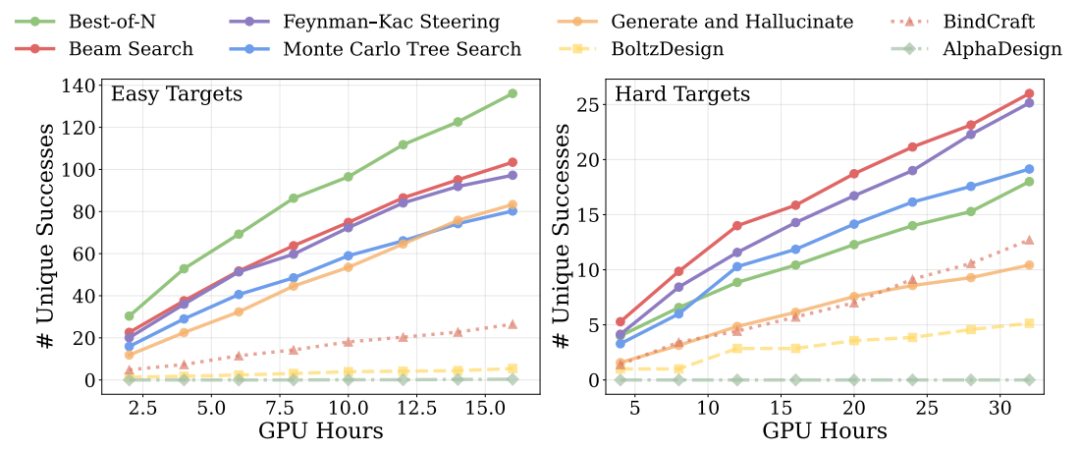
In computational extension experiments during the inference phase, the results show that on simple tasks, simply increasing the number of samples is enough to outperform all baseline methods, while on complex tasks, introducing more advanced search strategies (such as bundle search and Monte Carlo tree search) further expands the advantage.This indicates that model performance can continue to improve as the computational budget increases.As shown in the figure below
Regarding physical plausibility, further research is needed to optimize interfacial hydrogen bonds and related energy indices.The model was found to not only generate structurally sound binders, but also to optimize them at the fine-grained interaction level.This improves the stability of the bond.
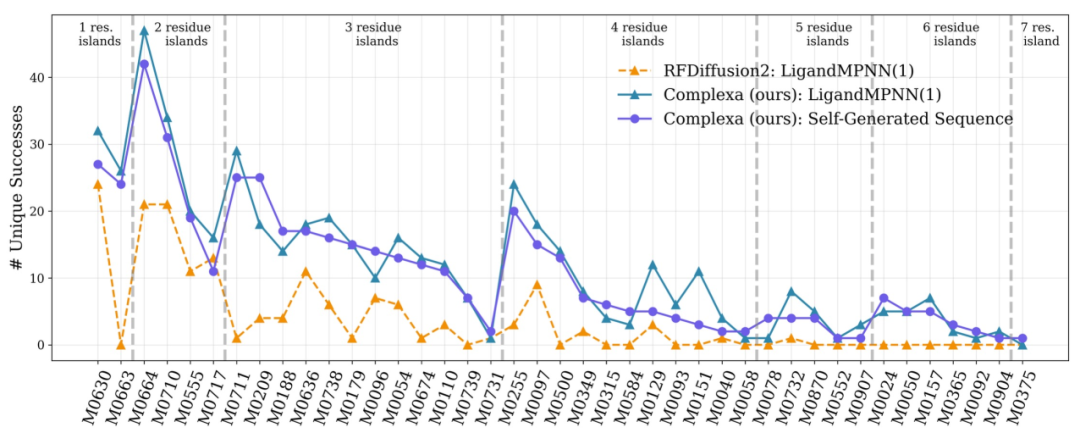
In more challenging multi-chain target tasks, existing methods cannot obtain efficient solutions under limited computational budgets.Complexa successfully generated high-quality candidates after expanding computing resources, demonstrating its scalability in complex problems.Finally, the tests on different tasks such as enzyme design are shown in the figure below. The framework has good generalization ability and can be extended from binder design to a wider range of protein engineering problems.
Paradigm Shift in AI Protein Design
In recent years, AI-driven protein binding agent design has been rapidly moving from theory to practice, and Nobel laureate David Baker and his team remain key leaders in the field. In 2025, their team published several studies in Science.The system verified the feasibility of designing highly specific pMHC binders based on RFDiffusion.The related work targeted 11 disease categories, successfully generated binding proteins that can drive T cells to recognize tumors, and verified the design accuracy at the atomic scale using cryo-electron microscopy, marking the beginning of verifiability for AI design.
Meanwhile, the MIT team explored a more integrated approach in the BoltzGen model, unifying structure prediction and aggregate generation into a single all-atom model and using continuous geometric representations instead of traditional discrete modeling.In experiments targeting 26 targets, 66% achieved nanomolar affinity as a binder.It also maintains a high success rate on out-of-distribution targets, demonstrating a certain generalization ability.
The industry is more focused on the engineering implementation of these capabilities. In early 2026, Bayer and Cradle entered into a three-year collaboration to integrate their AI protein engineering platform into the antibody development process. This platform has been applied in over 50 projects, significantly shortening development cycles and supporting a closed-loop iterative process of "design-test-learning." This signifies that AI is transitioning from an auxiliary tool to a fundamental capability within the R&D process.
Overall, the competition in protein design is shifting from single-model performance to system-level efficiency and scalability. Academia continues to push the boundaries of model capabilities, while industry is driving it into a stable and reusable R&D process. AI protein design is thus entering a more practical phase: the key is no longer "whether it can be designed," but "whether it can be designed continuously and efficiently."
Reference Links:
1.https://news.bioon.com/article/00bf92186439.html
2.https://mp.weixin.qq.com/s/1zKXUQtXgCJ7GA1_OUEShg
3.https://www.bayer.com/en/us/news-stories/ai-enabled-antibody-discovery-and-optimization