Command Palette
Search for a command to run...
MIT Has Proposed VibeGen, the First end-to-end Dynamic Protein Generation Model, Which Achieves a Bidirectional Mapping Between Sequence and vibration.
Proteins are the core functional molecules in living systems, but their functions are not determined solely by their static structure; rather, they originate from their constantly changing conformational dynamics. Within the framework of a complex energy landscape, proteins maintain dynamic equilibrium under physiological conditions through multi-scale movements ranging from femtoseconds to milliseconds, making them true molecular machines.
This is why abnormal protein dynamics are closely associated with a variety of diseases. For example, the tumor suppressor protein p53 functions in a manner dependent on conformational plasticity, and oncogenic mutations weaken this ability; CFTR mutations, on the other hand, induce cystic fibrosis by disrupting gating dynamics. These facts indicate that...The "movement" of proteins is itself an important determinant of their function.Therefore, understanding and designing proteins from a dynamic perspective is becoming a cutting-edge direction in structural biology and bioengineering.
Over the past few decades, researchers have developed experimental techniques such as nuclear magnetic resonance (NMR), hydrogen-deuterium exchange mass spectrometry (HDEMS), and cryo-electron microscopy (cryo-EM), as well as computational methods such as molecular dynamics simulations and vibrational normal modes (VMS) analysis to characterize protein dynamics. However, these methods are either too complex to scale up or too computationally expensive and time-limited, making them unsuitable for large-scale studies.
In recent years, deep learning and generative AI have brought new possibilities to protein research. Models such as AlphaFold2 have achieved high-precision structure prediction, and methods can also predict secondary structures, binding sites, and even vibrational features. However,Most existing methods still remain at the level of "structure or single property", lacking systematic modeling of intrinsic dynamics.In the design field, frameworks such as RFdiffusion and AlphaFold3 still treat structures as approximate rigid bodies and have not yet truly introduced dynamic constraints. Therefore, how to establish a unified mapping of "sequence-structure-dynamics-function" and achieve controllable design based on dynamics remains a core challenge.
Recently,A joint research team from MIT and Carnegie Mellon University proposed VibeGen, a protein-generating intelligent agent.By combining sequence generation with vibrational dynamics prediction, de novo protein design was achieved. The results show that proteins designed by this generative agent can not only fold into stable and novel structures, but also reproduce the distribution characteristics of target vibrational amplitudes at the main chain level.
The related research findings, titled "VibeGen: Agentic end-to-end de novo protein design for tailored dynamics using a language diffusion model," have been published in Matter.
Paper address:
https://www.cell.com/matter/abstract/S2590-2385(26)00069-X
Protein dynamics database based on low-frequency normal vibrational modes
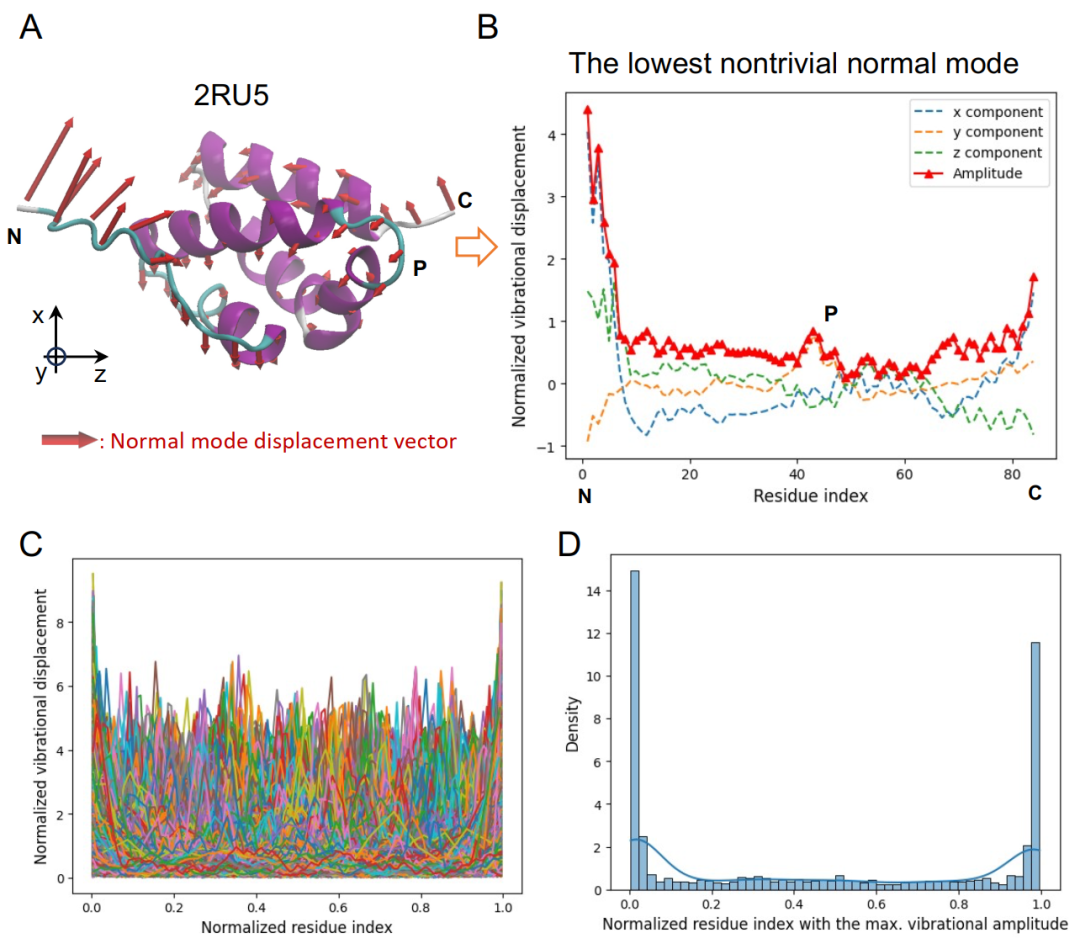
To build the database,Researchers screened single-chain proteins with a length of no more than 126 amino acids from the Protein Database (PDB) updated in January 2024.The structure was cleaned and completed using tools such as VMD, MMTSB, and SCWRL4. Energy minimization was then performed based on the CHARMM force field, and modal information was calculated using the block normal vibration mode method. After removing the first six rigid body modes representing overall translation and rotation, the lowest frequency nontrivial mode was selected for subsequent analysis.
Building upon this, the study further extracted the displacement modes of Cα atoms at each residue in the main chain, constructing a normal vibrational mode shape vector. The results showed a distinctly heterogeneous distribution of vibrational displacements: larger amplitudes at the chain ends and in loosely structured regions, while vibrations were restricted in dense regions such as α-helices and β-sheets. Turning and coiled regions exhibited local peaks due to their greater flexibility. To eliminate the influence of length differences, the vectors were normalized, making them coordinate-independent dynamic descriptors.
final,Researchers constructed a dataset containing 12,924 protein single strands.Analysis shows that the low-frequency vibration modes are highly diverse, with peak amplitudes concentrated at the chain ends. The dataset was divided into training and testing sets in a 9:1 ratio for subsequent training and evaluation of the generative model.
VibeGen: End-to-End De novo Protein Design Based on a Language Diffusion Model
The core challenge of this study is that the shape of normal vibrational modes is determined by the complex three-dimensional structure and elastic properties of proteins, and there is no direct mapping relationship between the sequence and the dynamics. At the same time, single-mode information is highly degenerate, and different sequences may correspond to similar dynamic characteristics, which makes the inverse design problem particularly difficult.
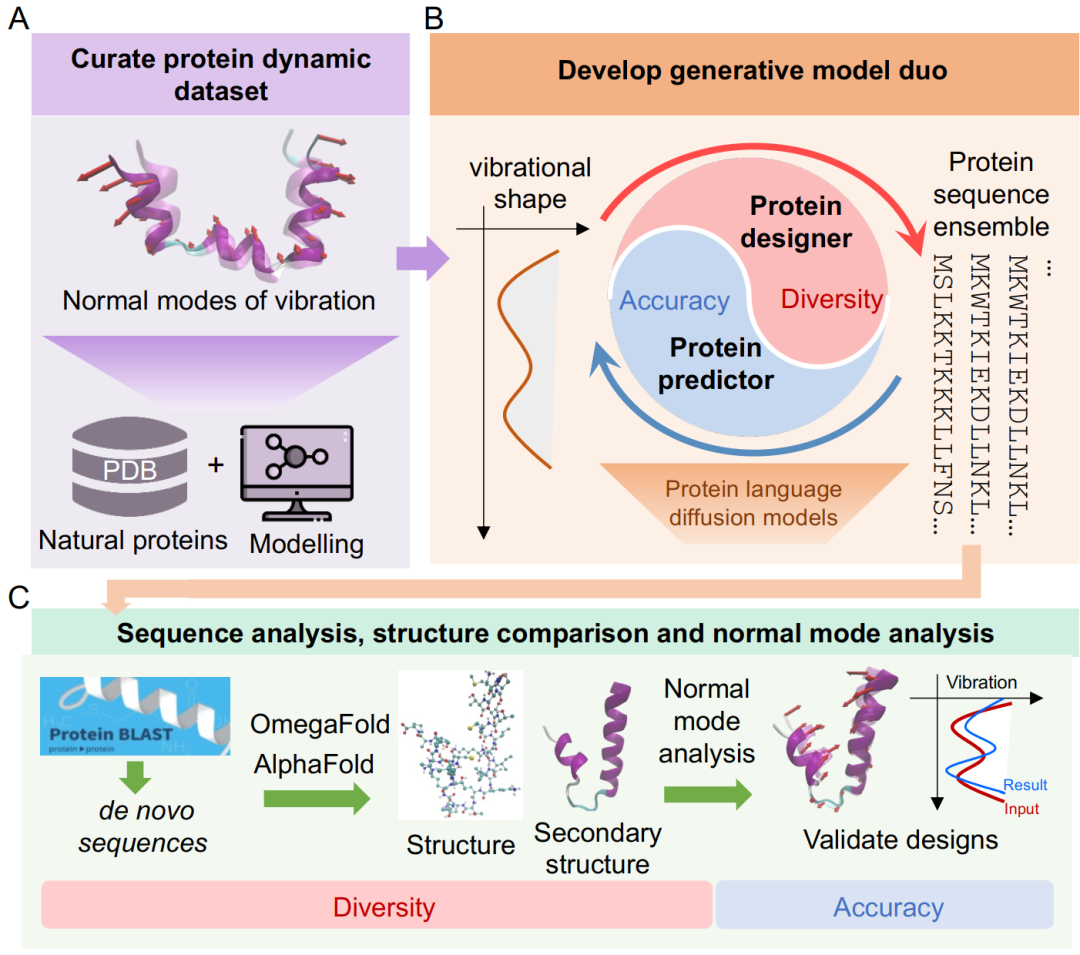
To address these challenges, this study first extracted key dynamic features of a large number of proteins from a protein database (PDB) through normal vibrational mode analysis and all-atom molecular dynamics simulations. Based on this,Researchers constructed two collaborative protein language diffusion models: a protein design module (PD) and a prediction module (PP).They are responsible for forward prediction and inverse design between the sequence and the normal vibrational mode space, respectively. The two modules have similar structures, both based on a combination of a pre-trained protein language model (pLM) and a diffusion model.
The task of the design module is to generate a sequence based on the target dynamic characteristics.During the denoising process, the diffusion model incorporates dynamic condition information through multiple channels and gradually generates sequences that conform to the target characteristics in the latent space.The prediction module has a symmetrical structure and infers the shape of the normal vibration mode from the input sequence. It also uses multiple sequence representations output by the pre-trained language model to optimize the prediction results.
The two modules are trained independently, and together they form a closed-loop collaborative system of "generation-evaluation-screening" during the deployment phase.The design module first generates candidate sequences, and the prediction module evaluates their dynamic performance in real time.Researchers can filter results based on their needs for accuracy or diversity, and repeat the iterations as necessary until a satisfactory sequence is obtained.
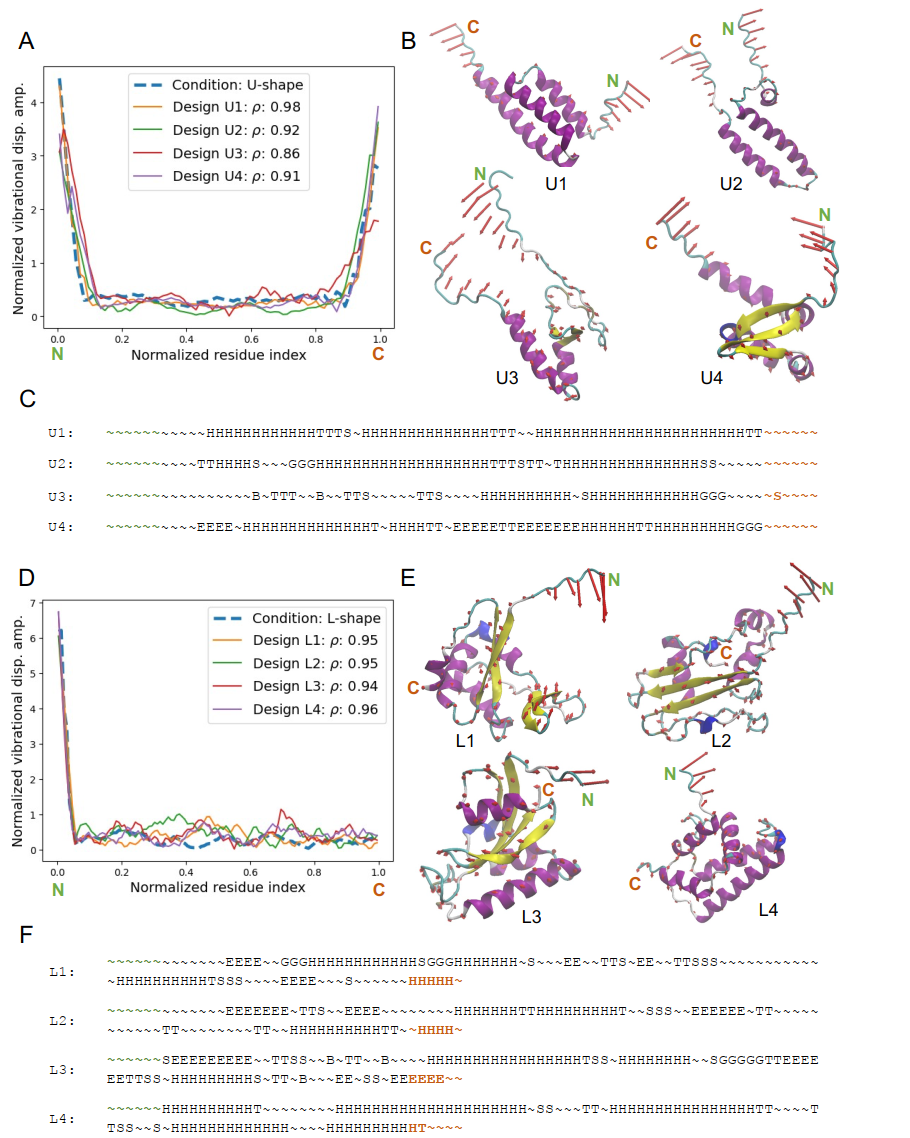
The model's performance was validated on the test set. For various typical normal vibrational mode shapes, including L-shaped, U-shaped, and W-shaped, the protein generated by the model was validated by actual normal vibrational mode analysis, and its vibrational shape highly matched the design target. Quantitative indicators such as Pearson correlation coefficient and relative L2 error showed that...This method can achieve high-precision design under complex dynamic constraints.
From a structural perspective, the formation of proteins exhibits a clear kinetic correspondence: regions with stronger vibrations tend to be random coils or flexible fragments, while regions with restricted vibrations tend to form stable structures such as α-helices or β-sheets.This demonstrates that the model has effectively captured the intrinsic relationship between structure and dynamics.
At the model implementation level, both the design and prediction modules use a medium-sized pre-trained model with 150 million parameters from the ESM-2 series as the pLM to balance computational efficiency and model performance. The diffusion model integrates conditional information into the denoising process through multiple channels of a U-shaped network and is independently trained using the Adam optimizer.
A breakthrough in both precision and novelty
To evaluate model performance, the study conducted experimental analyses across multiple dimensions. Diversity analysis showed that...For the same dynamic objective, the model can generate multiple design schemes with different structures but the same function.Taking U-shaped and L-shaped normal vibrational modes as examples, the designed proteins all exhibit a "dense core + open ends" layout: the ends are random coil structures, corresponding to high-amplitude regions; the core can be achieved in various ways, such as α-helical bundles or helical-fold hybrid structures, corresponding to low-amplitude regions. This diversity mainly comes from the degree of freedom in structural selection in the low-vibrational regions, and the model successfully captures and utilizes this "multiple solutions".
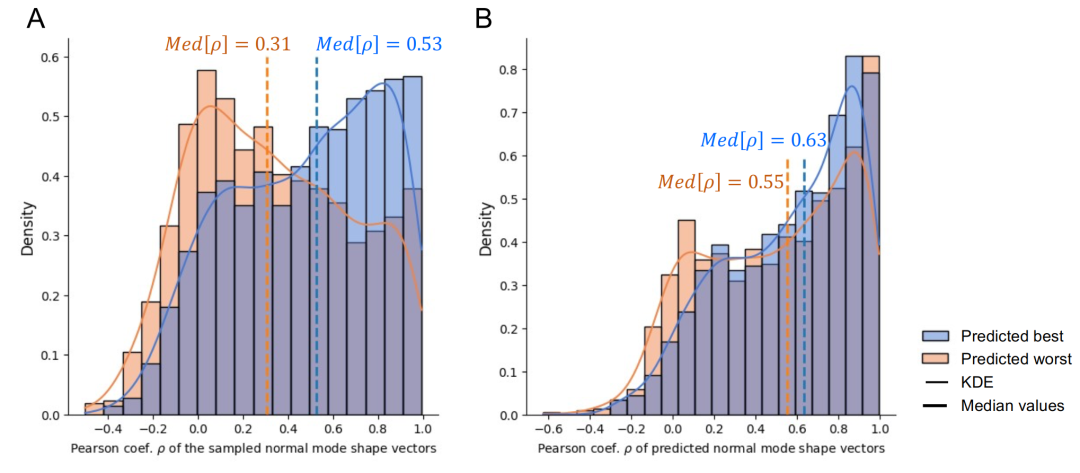
The effectiveness of the prediction module was verified through comparative experiments. As shown in the figure below, when selecting the best and worst prediction groups from the same set of candidate sequences, the actual design accuracy of the former was significantly higher than that of the latter (median Pearson correlation coefficient 0.53 vs 0.31), while the prediction module maintained stable prediction accuracy for both groups. This indicates that...Introducing a prediction module during the design process can effectively screen high-quality sequences and reduce reliance on expensive physical verification.
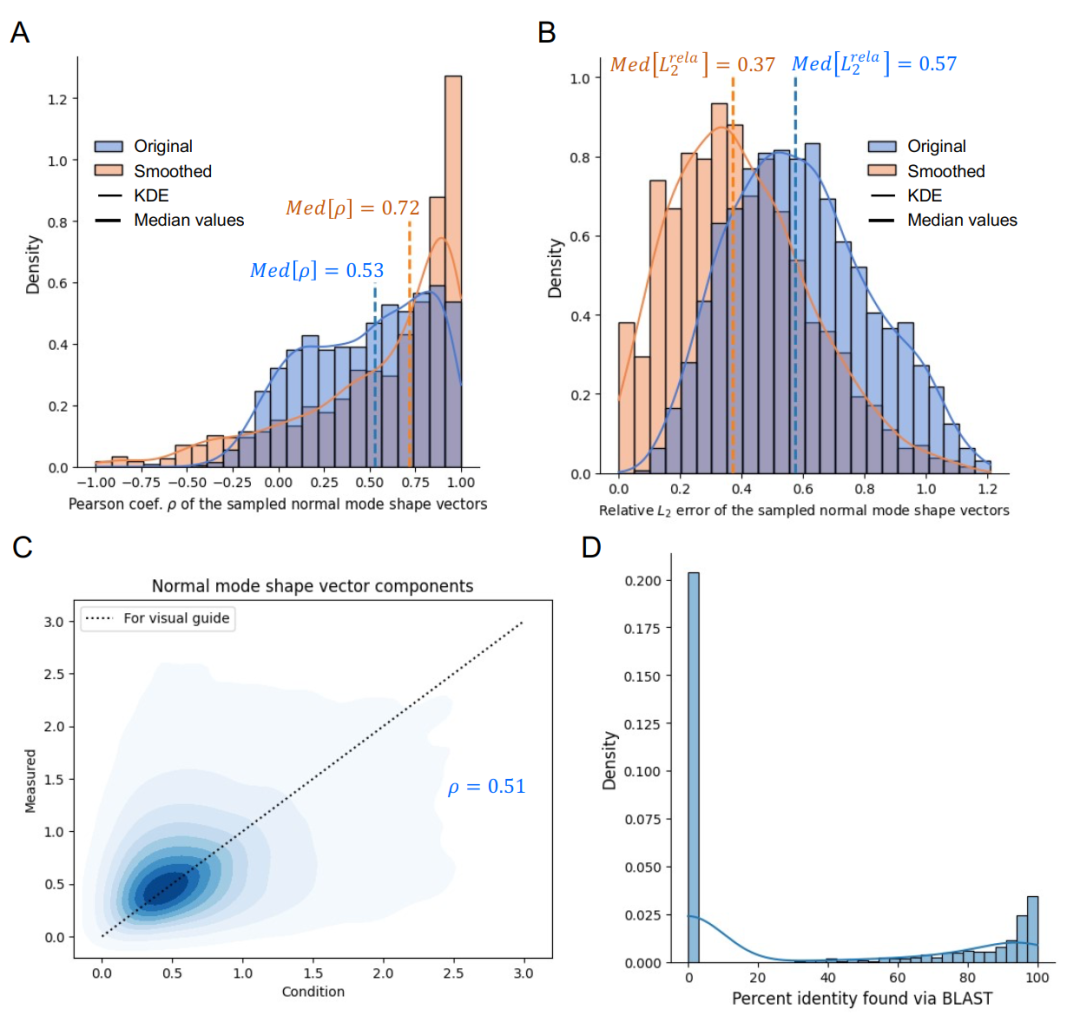
Overall performance statistics are based on 1,293 test cases. As shown in the figure below, the median correlation coefficient between the measured normal mode shape and the design target is 0.53, and the median relative L2 error is 0.57, reflecting the inherent difficulty of high-precision design at the residue level. After low-pass filtering to preserve the overall shape, the median correlation coefficient increases to 0.72, and the median error decreases to 0.37.This indicates that the model performs particularly well in capturing the overall vibration profile.This feature has the most important biological significance for the large-scale conformational dynamics of proteins.
In terms of novelty, the highest sequence identity of BLAST exhibits a bimodal distribution, with the main peak corresponding to de novo designed sequences.This indicates that the model is more likely to generate novel sequences, effectively expanding the potential library of protein structure and dynamics solutions.
The correlation between structure and dynamics is consistently shown in multiple sets of experiments: dense structures such as α-helices and β-folds are mostly distributed in low-amplitude regions, while high-amplitude regions are mostly loop regions or end curls.The model successfully captured this physical law and was able to control local flexibility with the help of secondary structural elements, demonstrating an understanding of the structure-dynamic relationship.
Overall, this model achieves a good balance of accuracy, diversity, and novelty in protein design under kinetic constraints, laying the foundation for subsequent more complex functional designs.
Combination of intelligent agent protein generation and normal vibrational mode inverse design
Research on intelligent agent protein generation and inverse design based on normal vibrational mode shapes is becoming a cutting-edge hot topic in the field of protein engineering, driving both academic exploration and industrial innovation.
In academia, numerous university teams have been continuously researching this area and have achieved a series of groundbreaking results. Some teams have optimized the intelligent agent collaborative framework,Combining normal vibrational mode analysis with a more advanced protein language diffusion model effectively alleviates the degeneracy problem in inverse design.This work further verified the intrinsic relationship between the shape of the normal vibrational modes and the secondary structure and dynamic properties of proteins, providing a more solid theoretical support and technical path for de novo design of proteins with specific functions.
Another team focused on lightweighting and generalization, optimizing the parameter size and training strategy of pre-trained protein language models, and developing smaller models that are easier to generalize.Furthermore, the application of inverse design of normal vibrational modes has been extended to specific fields such as enzyme catalytic site design and protein binding agent optimization.This has laid a solid foundation for subsequent industrial transformation.
In addition, Google DeepMind launched AlphaProteo,As the first artificial intelligence tool for designing novel high-strength protein adhesives, it can generate new protein conjugates for a variety of target proteins.Including vascular endothelial growth factor A, which is associated with complications of cancer and diabetes, the test achieved a higher experimental success rate. Its binding affinity is 3 to 300 times that of the best existing methods, which is expected to accelerate the development of anti-cancer and antiviral drugs, and also provides new ideas for the development of biosensors and the improvement of crop insect resistance.
Other companies are focusing on the pain points of drug development and using normal vibrational mode shape inverse design technology to design protein drugs for specific disease targets, shortening the development cycle, reducing costs, and promoting the development of protein drugs in a more precise and efficient direction.
Currently, the academic community's continuous optimization of design accuracy and model generalization ability, along with the industry's ongoing efforts to expand implementation efficiency and application scenarios, are jointly driving protein design technology towards greater precision, efficiency, and diversity. In the future, as the technology matures, protein design methods based on intelligent agents and normal vibrational mode analysis are expected to achieve wider applications in fields such as pharmaceuticals, industrial production, and biomanufacturing, bringing new breakthroughs.








